Case
46 Diagnosis |
Versión
en Español |
|||
Go back to clinical information and images
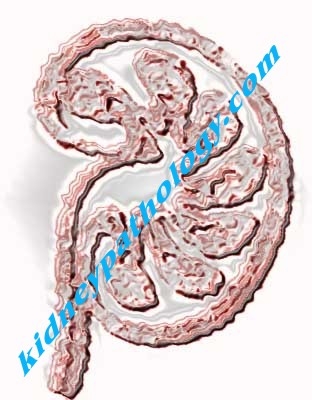
Diagnosis: Autosomal recessive polycystic kidney disease
Autosomal recessive polycystic kidney disease and congenital hepatic fibrosis (ARPKD/CHF) is an inherited disease characterized by non-obstructive fusiform dilatation of the renal collecting ducts leading to enlarged spongiform kidneys and ductal plate malformation of the liver resulting in congenital hepatic fibrosis. ARPKD/CHF has a broad spectrum of clinical presentations involving the kidney and liver.
ARPKD is a serious genetic disease characterized by cystic changes in the collecting ducts of the kidney and bile ducts within the liver. The gene for ARPKD (PKHD1) is located on chromosome 6p12 and encodes a protein called fibrocystin/polyductin (FPC), one of many proteins that are normally present at the primary cilia of the renal tubules and intrahepatic bile ducts. The severity of the clinical disease depends on the type of genetic mutations. Although exact function of FPC is not fully known, it is generally felt that like many of the other ciliary proteins, it plays a vital role in maintaining the structural integrity of organs such as kidney and liver, by modulating important cellular functions, including proliferation, secretion, apoptosis, and terminal differentiation. FPC probably works in conjunction with cellular proteins involved in autosomal dominant polycystic kidney disease that is, polycystin-1 and polycystin-2, which are also located in the primary cilia. Genetic abnormalities in PKHD1 may result in structural and functional abnormalities of FPC, leading to cystic phenotype (Al-Bhalal L, Akhtar M. Adv Anat Pathol. 2008 Jan;15(1):54-8. [PubMed link])
ARPKD classically presents antenatally or in the neonatal period; there are many cases of antenatal death. Later presentations do occur, however, in infancy or in adulthood. Neonates and infants with the classic phenotype have characteristic Potter's sequence, bilateral massive kidney enlargement and hepatic fibrosis.
ARPKD is characterized by pathologic changes in the kidney and/or liver. In the kidney, epithelial hyperplasia occurs along the collecting duct of the nephron. The hyperplastic cells undergo a functional change from being resorptive to becoming secretory. The fluid secreted from these abnormal cells is rich in epithelial growth factors, which further stimulate epithelial proliferation. The combination of epithelial hyperplasia and fluid secretion results in significant ductal ectasia. Approximately 10-90% of the ducts may be affected, resulting in a wide variability of renal dysfunction. Depending on the number of ducts involved, the kidneys may be massively enlarged. Examination of the kidney reveals multiple small subcapsular cystic spaces that correspond histologically with radially oriented, ectatic collecting ducts. It is very important in the differential diagnosis to know that in ARPKD there are not glomerular cysts. Liver disease is present in every patient with ARPKD, with the manifestations varying according to the patient's age at presentation. The chief pathologic hallmarks of liver disease are periportal fibrosis and biliary duct ectasia. Significant liver involvement is referred to as congenital hepatic fibrosis (Young BY, et al.In: E-medicine: http://emedicine.medscape.com/article/377154-overview Visited: December 18th, 2009).
ARPKD results in symmetrically enlarged kidneys that maintain their reniform shape. Beneath the capsule are scattered opalescent cysts from dilated collecting ducts, usually 1-2 mm in diameter but sometimes larger. On sections, the renal parenchyma resembles a sponge with ectatic, nonobstructed, radially oriented collecting tubules that have areas of hyperplastic cuboidal or low columnar lining epithelium. Interstitial fibrosis develops, but the glomeruli remain normal.
Since molecular diagnostics are not currently a feasible clinical tool for the diagnosis of most cystic kidney diseases, physicians must rely upon their clinical acumen and knowledge base in order to identify these patients.
See the chapter [Cystic renal diseases and develovemental defects] with atlas and text (this chapter is at the present only in Spanish).
Go back to clinical information and images
Bibliography
-
Denamur E, Delezoide AL, Alberti C, Bourillon A, Gubler MC, Bouvier R, Pascaud O, Elion J, Grandchamp B, Michel-Calemard L, Missy P, Zaccaria I, Le Nagard H, Gerard B, Loirat C; the Société Française de Foetopathologie. Genotype-phenotype correlations in fetuses and neonates with autosomal recessive polycystic kidney disease. Kidney Int. 2009 Nov 25. [Epub ahead of print] [PubMed link]
-
Turkbey B, Ocak I, Daryanani K, Font-Montgomery E, Lukose L, Bryant J, Tuchman M, Mohan P, Heller T, Gahl WA, Choyke PL, Gunay-Aygun M. Autosomal recessive polycystic kidney disease and congenital hepatic fibrosis (ARPKD/CHF). Pediatr Radiol. 2009 Feb;39(2):100-11. [PubMed link]
-
Rohatgi R. Clinical manifestations of hereditary cystic kidney disease. Front Biosci. 2008 May 1;13:4175-97. [PubMed link]
-
Young BY, Perlmutter S, Smith TH. Autosomal Recessive Polycystic Kidney Disease. In: E-medicine: http://emedicine.medscape.com/article/377154-overview Visited: December 18th, 2009.
-
Al-Bhalal L, Akhtar M. Molecular basis of autosomal recessive polycystic kidney disease (ARPKD). Adv Anat Pathol. 2008 Jan;15(1):54-8. [PubMed link]
-
Deane JA, Ricardo SD. Polycystic kidney disease and the renal cilium. Nephrology (Carlton). 2007 Dec;12(6):559-64. [PubMed link]
-
Adeva M, El-Youssef M, Rossetti S, Kamath PS, Kubly V, Consugar MB, Milliner DM, King BF, Torres VE, Harris PC. Clinical and molecular characterization defines a broadened spectrum of autosomal recessive polycystic kidney disease (ARPKD). Medicine (Baltimore). 2006 Jan;85(1):1-21. [PubMed link]
-
Shneider BL, Magid MS. Liver disease in autosomal recessive polycystic kidney disease. Pediatr Transplant. 2005 Oct;9(5):634-9. [PubMed link]