
Enfermedades quísticas renales y defectos del desarrollo
Las enfermedades quísticas que afectan al riñón pueden ser hereditarias o adquiridas e incluyen: Enfermedad poliquística autosómica dominante y autosómica recesiva; riñón en esponja medular; complejo enfermedad quística medular-nefronoptisis familiar; enfermedad glomeruloquística; enfermedad quística lozalizada; quistes simples; quistes renales en esclerosis tuberosa y en enfermedad de von Hippel-Lindau; quiste renal multilocular; y enfermedad quística adquirida (asociada a diálisis). Aquí revisaremos los hallazgos anatomopatológicos característicos y los aspectos patogénicos y clínicos más relevantes. En una segunda parte (en esta misma página) mencionaremos algunas de las malformaciones (defectos del desarrollo) más frecuentes: agenesia renal, hipoplasia, adisplasia renal hereditaria, displasia renal, nefromegalia congénita, oligomeganefronia (hipoplasia oligonefrónica), disgenesia tubular renal, y riñón en herradura (fusión renal).
ENFERMEDADES QUÍSTICAS RENALES
La forma más común de quistes renales son los quistes simples y la enfermedad quística adquirida (generalmente asociada a diálisis). En la patogénesis de la formación de quistes renales juega un papel importante la obstrucción de túbulos, que se distienden y se hacen quísticos debido al flujo continuo de orina, sin embargo, este no es el único mecanismo involucrado en su génesis. Se han mencionado tres aspectos fundamentales para su desarrollo: 1.) proliferación anormal y/o carencia de diferenciación de las células epiteliales renales; 2.) fluido continuo de líquido (orina); y 3.) anormalidades en la membrana basal tubular y/o matriz extracelular (Kern WF, et al. Atlas of Renal Pathology. W.B. Saunders Company, Philadelphia, 1999, p. 235).
ENFERMEDAD POLIQUÍSTICA RENAL AUTOSÓMICA DOMINANTE (EPRAD)
Ha sido conocida también como enfermedad poliquística del adulto, pero el primer nombre es el más adecuado ya que es una enfermedad hereditaria y que puede, potencialmente, diagnosticarse en la niñez. Su incidencia en la población general es de alrededor de 1 por 1.000 y se cree que afecta más a personas de raza blanca. Es la tercera causa más frecuente de falla renal terminal, aproximadamente 6% - 10% de pacientes en diálisis y trasplante tienen EPRAD.
Las alteraciones genéticas en la EPRAD son heterogéneas y se han descrito varios loci, el más común (aproximadamente el 85% de casos) está localizado en el cromosoma 16p13.3: PKD1 (Reeders et al, Nature 317:542-4, 1985 [PubMed link]); otro loci se ha identificado en 4q12-22: PKD2; y hay algunas familias con EPRAD sin alteraciones en el PKD1 ni PKD2, por lo que deben haber otros genes involucrados. En los casos de familias afectados por mutaciones en PKD2 el curso de la enfermedad tiende a ser más indolente. En un porcentaje importante de casos (hasta el 25%) no hay antecedentes familiares de EPRAD, estos casos podrían deberse a fallas en identificar pacientes con la enfermedad o a mutaciones espontáneas. Otro factor que parece influir en este hallazgo es el grado de expresión de las alteraciones, al parecer, en algunas personas no se desarrolla completamente la enfermedad y algunos casos son asintomáticos aun en edades avanzadas. Otros autores consideran que hay una penetrancia genética del 100% a los 80 años de edad. Lo que parece claro es que el tipo de alteración genética es variable y hay heterogeneidad en la expresión clínica (Fencl F, et al. Genotype-phenotype correlation in children with autosomal dominant polycystic kidney disease. Pediatr Nephrol. 2009;24:983-9. [PubMed link])
El gen PKD1 codifica una proteína llamada policistina, una proteína transmembrana con múltiples dominios funcionales y, probablemente, con un papel en la adhesión celular y en la interacción células-matriz, siendo también importante en la maduración celular. La proteína PKD2 tiene considerable homología con la policistina. El mecanismo por el cual la alteración genética lleva a la producción de quistes y daño renal no está muy bien entendido; al parecer, el menos en muchos casos, hay proliferación de células epiteliales y bloqueo en la maduración o diferenciación de ellas. La hiperplasia epitelial resultante sería origen (o cofactor) en la dilatación de túbulos y, en alguna mediada, en su obstrucción. Además, suele haber un aumento de la presión hidrostática del fluído dentro del quiste. Defectos en la elasticidad de la membrana basal tubular podrían ser otro factor clave en el desarrollo de la enfermedad. De hecho, en algunos pacientes con EPRAD hay también quistes en otros órganos, aneurismas intracraneales, divertículos de colon, dilatación de segmentos de aorta y prolapso de la válvula mitral.
Clínica: La presentación clínica más clásica es dolor abdominal bajo que empeora con el ejercicio físico, azoemia, masa palpable en flancos e historia familiar de ERPAD en un paciente en la tercera década de la vida o mayor. El diagnóstico puede confirmarse por estudios de imagen. Es común la hematuria micro o macroscópica y proteinuria leve; en muchos pacientes hay hipertensión arterial sistémica y anemia; en poco menos de un 20% de pacientes hay cálculos en vías urinarias. La falla renal se instaura progresivamente; al comienzo puede haber sólo una disminución en la capacidad de concentración de la orina. Con frecuencia hay infecciones urinarias agregadas. Ocasionalmente se informa de diagnóstico de la enfermedad en niños (con alteraciones quísticas en la infancia) pero usualmente cuando tienen historia familiar de la enfermedad.
Hay considerable variabilidad fenotípica que puede reflejar mutaciones diferentes en el mismo gene, mutaciones en diferentes genes o la influencia de otros factores genéticos o medioambientales. Hay notoria similaridad en la edad de presentación y progresión a falla renal en diferentes miembros de una misma familia.
En el hígado suele haber también múltiples quistes, variando mucho entre casos. El órgano suele estar aumentado de tamaño pero no hay alteración de su función. En general son pequeños, afectan conductos porta, tienen pared delgada y fibrosa y estan tapizados por epitelio biliar; no comunican con conductos biliares normales. Se han descrito casos de fibrosis portal difusa, aunque es excepcional. Otros órganos en los que se han descrito quistes son: páncreas, bazo, tiroides y vesículas seminales. Hay divertículos de colon en hasta el 80% de pacientes; aneurismas de la arteria cerebral en una tercera parte de casos; aneurismas aórticos; dilatación de la raíz aórtica; y enfermedad de la válvula mitral.
Macroscópicamente hay un marcado aumento de ambos riñones con cambios quísticos difusos. Los quistes están distribuidos en todo el riñón (médula y corteza), con pérdida completa de su anatomía y poco estroma entre ellos. Estos quistes suelen ser uniloculados, de tamaño muy variable: desde milímetros a varios centímetros, no comunican entre sí y contienen líquido claro, amarillo, ocre, marrón o con contenido gelatinoso; en algunos quistes hay hemorragia. En pacientes en falla renal suele haber compromiso difuso, bilateral y simétrico, pero, en estadios más iniciales es menos homogénea la distribución, incluso, puede confundirse con quistes simples múltiples. El peso de cada riñón está marcadamenmte aumentado, con una media de 2.600 g.
Figura 1. Aspecto carcaterístico de riñón con EPRAD. El órgano está muy aumentado de tamaño y completamente reemplazado por cavidades quísticas de tamaño variable, no comunicantes entre sí ni con la pelvis renal, , con paredes delgadas y contenido claro, amarillo o café. Este aspecto macroscópico permite confirmar el diagnóstico en muchos de los casos.
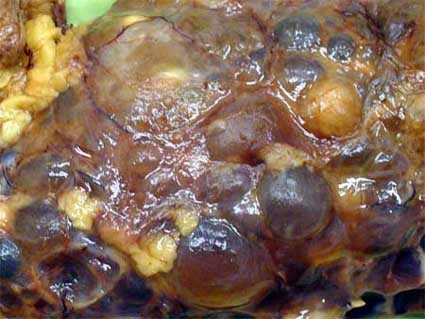
Figura 2. Los quistes tienen pared translúcida, contenido claro y paredes delgadas. Protruyen en la superficie del órgano y le dan un aspecto lobulado.
Histopatología
Los quistes están tapizados por una sola capa de células epiteliales con ocasionales focos de hiperplasia polipoide; el número de células incrementa en proporción al incremento del área superficial de los quistes. Las células están aplanadas y, aunque en la mayoría no es posible identificar el tipo del túbulo del cual surge el quiste, en algunos casos pueden identificarse células epiteliales de tipo glomerular parietal, tubular proximal o de conductos colectores. Ocasionalmente se identifican restos de penacho glomerular indicando su origen en la cápsula de Bowman. Siempre hay parénquima residual entre los quistes, un hallazgo histológico muy importante para diferenciarlo del quiste multilocular (o nefroma multiquístico); en este parénquima solemos encontrar cambios crónicos: glomeruloesclerosis, fibrosis intersticial y atrofia tubular.
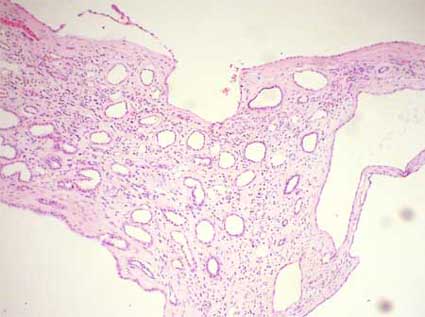
Figura 3. Los quistes están revestidos por epitelio tubular, con células cúbicas o aplanadas por compresión. El estroma entre los quistes es de tipo renal, con glomérulos y túbulos, generalmente, atróficos. Este hallazgo permite diferenciar la EPRAD de lesiones neoplásicas o pseudoneoplásicas multiquísticas como el quiste multilocular o el carcinoma multiquístico de células renales. (H&E, X200).
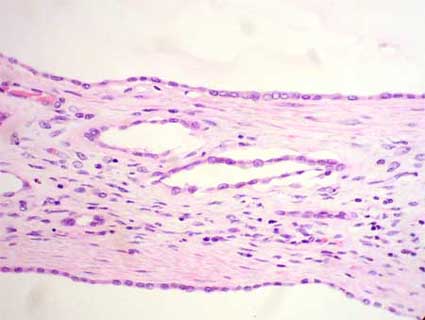
Figura 4. A mayor aumento se observa mejor el recubrimiento epitelial simple y el estroma renal entre los quistes. (H&E, X300).
Estudios de microdisección han demostrado que los quistes se pueden originar en cualquier parte de la nefrona o conductos colectores y, usualmente, conservan su conección con el túbulo de origen hasta que alcanzan tamaños grandes. Algunos autores sugieren que sólo 1 - 2% de las nefronas son quísticas (Grantham JJ, et al, Kideny Int 31:1145, 1987 [PubMed link])
En niños en los que se ha demostrado la enfermedad, los quistes suelen ser de origen glomerular y la ERPAD responde por la mitad de casos de la llamada enfermedad glomeruloquística (ver más adelante).
Por microscopía electrónica se demuestra pérdida de microvellosidades apicales e interdigitaciones celulares laterales; la membrana basal suele estar engrosada o multilaminada.
Diagnóstico diferencial
La presencia de múltiples quistes bilaterales en un paciente con historia familar establece el diagnóstico y la presencia de quistes en otros órganos como el hígado ayuda a confirmarlo. En casos de enfermedad leve/inicial puede confundirse con quistes simples múltiples, en estos casos necesitaremos de la historia familiar y/o detección de compromiso de otros órganos. En las formas autosómicas recesivas no suelen haber quistes hepáticos. La presencia de cartílago u otro tejido heterotópico y de parénquima de aspecto embrionario o fetal sugiere displasia renal, más que enfermedad poliquística hereditaria. Un carcinoma renal multiquístico o un quiste multiloculado (nefroma multiquístico) suele se unilateral y deja áreas amplias de tejido renal normal; además, entre los quistes no suele detectarse parénquima renal conservado.
ENFERMEDAD RENAL POLIQUÍSTICA AUTOSÓMICA RECESIVA (ERPAR)
También conocida como enfermedad poliquística infantil. Es poco frecuente: 1 en 6.000-55.000 nacimientos vivos. La enfermedad renal poliquística autosómica recesiva (ERPAR) es una enfermedad hereditaria caracterizada por dilataciones fusiformes no obstructivas de los conductos colectores renales produciendo riñones grandes con aspecto espongiforme (como esponjas) y malformaciones de la placa ductal del hígado que llevan a fibrosis hepática congénita. La ERPAR tiene un espectro amplio de presentaciones clínicas que comprometen riñones e hígado.
ERPAR es una enfermedad genética seria caracterizada por cambios quísticos en los conductos colectores renales y conductos biliares hepáticos. El gen de la ARPKD: PKHD1, esta localizado en el cromosoma 6p12 y codifica una proteína llamada fibrocistina/poliductina (FPC), una de las muchas proteínas que están normalmente presentes en los cilios primarios de los túbulos renales y conductos biliares intrahepáticos. La severidad de la enfermedad depende del tipo de mutación genética. Aunque la función exacta de la FPC no se conoce completamente, se creee que como muchas otras proteínas ciliares, juega un papel esencial en el mantenimiento de la integridad estructural de órganos como el riñón y el hígado, modulando importantes funciones celulares que incluyen proliferación, secreción, apoptosis y diferenciación terminal. La FPC probablemente trabaja en conjunto con otras proteínas celulares involucradas en la enfermedad renal poliquística autosómica dominante: policistina-1 y policistina-2, las cuales están localizadas en los cilios primarios. Anormalidades genéticas en PKHD1 pueden producir anormalidades estructurales y funcionales de la FPC que llevan a poliquistosis (Al-Bhalal L, Akhtar M. Adv Anat Pathol. 2008 Jan;15(1):54-8. [PubMed link])
La ERPAR suele presentarse antes del nacimiento o en el periodo neonatal; hay muchos casos de muerte intrauterina. En otros casos la presentación clínica puede ser más tardía, incluyendo casos que se diagnostican durante la edad adulta. Los neonatos y niños pequeños con el fenotipo clásico tienen características de la secuencia de Potter, aumento renal masivo bilateral y fibrosis hepática.
La enfermedad se caracteriza anatomopatológicamente por cambios en riñones y/o hígado. En riñones hay hiperplasia de células epiteliales en los conductos colectores. Las células hiperplásicas sufren un cambio funcional y en vez de ser reabsortivas son secretoras. El líquido de secreción de estas células es rico en factores de crecimiento epitelial, el cual además estimula la proliferación epitelial. La combinación de hiperplasia epitelial y abundante secresión produce una significativa ectasia ductal. Aproximadamente entre el 10% y el 90% (de acuerdo al fenotipo de la enfermedad) de los conductos colectores pueden estar afectados, resultando en una amplia variabilidad en el grado de disfunción renal. Dependiendo del número de conductos alterados los riñones pueden estar masivamente aumentados de tamaño o sólo tener incremento leve. El examen macroscópico de los riñones revela múltiples pequeños espacios quísticos subcapsulares (como se ven en la Figura 4B), que corresponden histológicamente a conductos colectores ectásicos orientados radialmente (perpendiculares a la cápsula). Es muy importante en el diagnóstico diferencial saber que en la ERPAR no hay quistes glomerulares (sí se evidencian en la enfermedad autosómica dominante y otras poliquistosis). Hay enfermedad hepática en todos los pacientes con ERPAR, aunque el grado de afectación es muy variable. Las alteraciones histológicas características en el hígado son la fibrosis periportal y la ectasia de conductos biliares (Figura 4E). En general, a mayor edad del paciente habrá menor compromiso renal y mayor compromiso hepático. El compromiso hepático significativo se conoce como "fibrosis hepática congénita" (Young BY, et al.In: E-medicine: http://emedicine.medscape.com/article/377154-overview Visitado: Diciembre 18 de 2009).
Dado que las pruebas moleculares no son en la actualidad una herramienta fácilmente disponible para el diagnóstico en la mayoría de casos, los médicos deben basarse para su diagnóstico en el conocimiento de esta enfermedad y agudeza clínica.
Clínica: La enfermedad se puede presentar a cualquier edad desde el nacimiento hasta adultos jóvenes, de allí que la denominación de enfermedad poliquística infantil no sea la más adecuada. En las formas más clásicas, reconocidas desde antes de los setenta, hay muerte prenatal o poco después del nacimiento debido a insuficiencia pulmonar en relación con el síndrome de Potter o con dificultad mecánica para respirar debido a la compresión por el tamaño de los riñones; hay oligohidramnios e hipoplasia pulmonar. El diagnóstico puede hacerse antes del nacimiento. En pacientes que sobreviven al período neonatal puede haber insuficiencia renal leve pero progresiva. Los riñones son palpables y tienden a crecer. La orina muestra proteinuria leve y baja densidad: es característico el defecto de concentración de la orina. La severidad de la anemia normocítica, normocrómica va paralela al grado de insuficiencia renal. Es frecuente la hipertensión arterial sistémica.
Las manifestaciones clínicas de los pacientes que no fallecen antes o inmediatamente después del nacimiento varían con la edad. Encontraremos enfermedad renal y hepática casi en todos, pero, la enfermedad renal tiende a dominar el cuadro en niños pequeños y la enfermedad hepática es la predominante en niños mayores y adolescentes. En algunos casos el daño hepático puede presentarse sin insuficiencia renal. En pacientes con afectación predominantemente hepática encontraremos hipertensión portal, esplenomegalia y sangrado por varices esofágicas; la función metabólica hepática está usualmente conservada.
Macroscópicamente, en casos de diagnóstico al nacimiento, los riñones están muy aumentados de tamaño, con distensión de la cavidad abdominal, con un peso conjunto de 200-600 g. El aumento renal usualmente es simétrico y suele conservarse el aspecto "reniforme" del órgano. La superficie capsular muestra múltiples quistes de 1-2 mm. Los riñones están comprometidos difusamente, hay quistes estrechos, elongados y orientados radialmente (perpendiculares a la cápsula) en la corteza; la médula también presenta numerosos quistes pero su orientación es más irregular. El aspecto del órgano, al corte, es espongiforme.

Figura 4A. Aspecto macroscópico de un riñón con ERPAR en un mortinato, Note la apariencia espongiforme generalizada, el aspecto radiado de las dilataciones quísticas y la sutil diferencia entre la corteza y la médula.

Figura 4B. La superficie externa del riñón poliquístico permite evidenciar los pequeños quístes subcapsulares que varían, usualmente, entre 1 y 2 mm. Estos corresponden a túbulos y conductos colectores dilatados que vienen desde la médula, por lo que dan un aspecto de dilataciones fusiformes radiadas (derecha). Acercamiento del mismo caso de la figura 4A.
En niños mayores, adolescentes o adultos jóvenes el aspecto es más variable, con menos quistes y más grandes, el tamaño de los riñones puede estar aumentado, ser más o menos normales o aun presentar disminución de él. En algunos casos el aspecto es similar al de la enfermedad autosómica dominante y en otros al del riñón en esponja medular.
Histopatología
En niños muy pequeños y en mortinatos: Parénquima casi completamente reemplazado por túbulos dilatados que aparecen elongados en la corteza y redondeados u ovales en la médula, dando a la corteza un aspecto diferente al de la médula; están predominantemente recubiertos por epitelio cúbico simple con focos de hiperplasia y proyecciones papilares; el epitelio es cúbico o columnar bajo. Los quistes están originados en conductos colectores; los túbulos proximales y glomérulos están comprimidos entre ellos. No hay quistes glomerulares. En algunos casos puede identificarse dilatación de túbulos distales y asas de Henle. Hay grados variables de fibrosis intersticial, la cual es progresiva con la evolución de la enfermedad, pero los glomérulos suelen tener aspecto normal.

Figura 4C. Múltiples quietes de tamño variavle, el epitelio es cilíndrico bajo, cúbico o puede verse plano por compresión. No hay quistes glomerulares. Entre los quístes hay parénquima renal que puede mostrar, en algunos casos, fibrosis. H&E, X100.

Figura 4D. Cavidades quísticas y túbulos dilatados, el epitelio puede verse hiperplásico. Note los glomérulos preservados, en este caso con aspecto fetal (el caso corresponde al de un mortinato), pero sin dilatación del espacio de Bowman. En casos de pacientes que llegan a la adolescencia o edad adulta podremos ver glomeruloesclerosis y fibrosis intersticial. H&E, X200.
En niños mayores, adolescentes y adultos: Hay menos quistes que comprometen menos áreas del parénquima y son más grandes, esféricos e irregulares. Hay cambios fibróticos y esclerosis glomerular en una extensión variable. En algunos casos el único hallazgo puede ser algunos conductos dilatados en la médula (principalmente adultos).
La fibrosis hepática congénita esta casi invariablemente acompañando las alteraciones renales. La lesión compromete espacios porta y consiste en incremento del tejido conectivo fibroso con aparente aumento de conductos biliares que se ven dilatados y, a veces, ramificados. Esta apariencia de conductos biliares puede representar una detención de la maduración normal. En el hígado u otros órganos no hay quistes.

Figura 4E. Hígado del mismo mortinato de la figura anterior. Se evidencia fibrosis en espacios porta, con aumento de conductos biliares los cuales aparecen ramificados. Estos hallazgos son descritos por algunos como complejos de von Meyenburg. Tricrómico de Masson, izquierda X100; derecha X200.
Diagnóstico diferencial
En casos de pacientes afectados en edades cercanas a la adolescencia o mayores puede planterase el diagnóstico de enfermedad autosómica dominante; en la EPRAR no hay quistes glomerulares, no hay quistes hepáticos y no se asocia a enfermedad autosómica dominante en su familia. En algunos casos con alteraciones predominantemente medulares se puede plantear el diagnóstico de riñón en esponja medular, pero, en esta última no hay fibrosis hepática congénita.
Se le ha llamado también ectasia renal tubular, enfermedad quística de las pirámides renales y ectasia canalicular precalicial. Se caracteriza por dilatación de conductos colectores en las pirámides renales, usualmente sin compromiso de la función renal. Se le ha considerado una anormalidad del desarrollo, aunque no suele detectarse en niños pequeños. La mayoría de casos son esporádicos y hay algunos informes de casos en los que se sugiere herencia autosómica dominante. La enfermedad ha sido asociada con hemihipertrofia congénita del cuerpo, síndrome de Ehlers-Danlos, síndrome de Marfan y síndrome de Wiedemann-Beckwith.
Clínica: Usualmente asintomática; se detecta por pielografía intravenosa u otros estudios de imagen en los que se demuestra estriaciones lineales o quistes esféricos en la papila renal y acumulación de medio de contraste en estos conductos quísticos o ectásicos. Ocasionalmente se ha asociado con hematuria, infecciones y litiasis.
Macroscópicamente hay pequeños quistes, de entre 1 y 5 mm, y conductos ectásicos en las pirámides; más prominentes cerca de la punta de la papila. Puede comprometer todas las papilas o unas pocas y es bilateral en el 75% de casos. La corteza y superficie renal tienen aspecto normal. A veces están disminuidos de tamaño debido a infecciones crónicas (pielonefritis). Muy pocas veces nos corresponde a los patólogos evaluar un especimen con esta enfermedad, dado que no necesita un tratamiento especial (menos aun nefrectomía).
Histopatología
Los quistes y conductos ectásicos están tapizados por epitelio columnar, por epitelio transicional estratifiacado u, ocasionalmente, por epitelio escamosos estratificado. En el intersticio de la médula usualmente hay fibrosis e inflamación crónica. Ocasionalmente hay pielonefritis aguda. En el resto del parénquima no hay cambios específicos. Ocasionalmente hay ulceración y depósitos de calcio en los quistes.
En el diagnóstico diferencial la principal consideración es con el complejo nefronoptisis - enfermedad quística medular; en esta última los quistes se localizan predominantemente en la unión cortico-medular.
COMPLEJO NEFRONOPTISIS - ENFERMEDAD QUISTICA MEDULAR
La enfermedad quística medular y la nefronoptisis juvenial (o nefronoptisis familiar) fueron inicialmente descritas como dos enfermedades diferentes; la primera se caracteriza por alteraciones quísticas predominantemente medulares y la segunda se describió como una nefropatía familiar autosómica recesiva. Sin embargo, las características patológicas y clínicas comunes y la presentación de casos con y sin quistes en pacientes de la misma familia han hecho desaparecer el límite entre ambas. Aunque las alteraciones patológicas pueden ser indistinguibles, hay una clara heterogeneidad hereditaria: formas autosómicas recesivas, autosómicas dominantes y esporádicas. En los casos de enfermedad autosómica recesiva se ha identificado un gen en el cromosoma 2q.
Clínica: En cerca de la mitad de los casos hay antecedentes familiares de enfermedad autosómica recesiva. Estos pacientes son niños con azoemia progresiva llevando a falla renal terminal en la segunda década de la vida. Ambos sexos se afectan igualmente. La enfermedad es responsable del 20 al 25% de niños que entran a diálisis crónica. Hay retraso en el crecimiento y poliuria y polidipsia en relación con alteración de la capacidad de concentración de la orina. En muchos casos hay pérdida de sal (que ayuda a proteger contra la hipertensión). En algunos hay proteinuria leve. En formas autosómicas recesivas hay anormalidades extrarrenales frecuentes: oculares, retinitis pigmentosa, fibrosis hepática congénita (raro) y anormalidades esqueléticas.
En pacientes con herencia autosómica dominante la presentación clínica suele ser en la edad adulta, no se suelen asociar con manifestaciones extrarrenales y la evolución a falla renal terminal es igualmente rápida.
Macroscópicamente los riñones están retraídos, disminuidos de tamaño, firmes y pálidos. Hay adelgazamiento de corteza y médula y, en muchos casos, quistes de pared delgada y con líquido en su interior, que se ubican cerca de la unión cortico-medular; su número es variable. En cerca del 25% de casos no se detectan. Su tamaño oscila desde menos de 1 milimetro hasta 1 cm. Se han identificado divertículos tubulares en asa de Henle, túbulos distales y conductos colectores.
Histopatología
Los cambios son inespecíficos. Los quistes se suelen encontrar cerca d la unión cortico-medular, están recubiertos por epitelio aplanado que es continuación del de los conductos colectores. En algunos túbulos adyacentes hay dilatación de su luz. No hay calcificaciones de la pared de los quistes, cálculos intraquísticos, ni compromiso de la papila como en el riñón en esponja medular.
En corteza y médula hay cambios crónicos túubulo-intersticiales y vasculares inespecíficos. En glomérulos hay esclerosis global y/o segmentaria.
En casos leves o iniciales el diagnóstico anatomo-patológico es prácticamnte imposible y debe basarse en los hallazgos clínicos y antecedentes familiares.
El término enfermedad glomeruloquística es usado para describir un grupo heterogéneo de desórdenes, más que para denominar una enfermedad específica. Los quistes glomerulares pueden encontrarse en enfermedad poliquística autosómica dominante (EPRAD), síndrome hepato-renal de Zellweger, esclerosis tuberosa, trisomía 13, síndrome oro-facial-digital (síndrome de Ochoa), etcétera. Este diagnóstico sólo debe aplicarse cuando el hallazgo histopatológico predominante es la dilatación quística de glomérulos.
Alrededor de la mitad de pacientes son jóvenes o niños con EPRAD. De los restantes, la mayoría tendrán otras alteraciones renales con formación de quistes glomerulares. Aun así, parece que hay algunos casos en los que ésta podría ser una enfermedad separada, más que una variante de EPRAD.
La enfermedad renal glomerulocística (GCKD, por sus siglas en inglés) es una forma poco frecuente de enfermedad hereditaria caracterizada por la dilatación quística de la cápsula de Bowman y porción inicial del túbulo contorneado proximal. Los espacios de Bowman dilatados están recubiertos por un epitelio aplanado y contienen penachos glomerulares rudimentarios. La GCKD se define como una dilatación de dos a tres veces del espacio de Bowman en más del 5% de los glomérulos identificables. Los quistes glomerulares en la GCKD primaria se localizan principalmente en la región subcapsular del riñón (Rasouly HM, et al. Loss of Zeb2 in mesenchyme-derived nephrons causes primary glomerulocystic disease. Kidney Int. 2016;90(6):1262-1273. [PubMed link]).

Figura 4F. Quistes glomerulares en un caso de enfermedad renal glomeruloquística hereditaria (Caso 157 de nuestra serie de casos). Todos los glomérulos de la biopsia presentaban este aspecto y no había dilataciones quísticas en túbulos. Tricrómico de Masson, aumento original, X100.
En las imágenes ecográficas, los riñones pueden aparecer agrandados, con un aumento de la ecogenicidad y pérdida de la diferenciación corticomedular. La GCKD familiar suele identificarse con riñones hipoplásicos o de tamaño normal. Sin embargo, tanto en formas esporádicas como familiares, los riñones pueden ser hipoplásicos, de tamaño normal o agrandados. Pueden evidenciarse quistes puntiformes que ayudan a distinguir esta alteración de la enfermedad renal poliquística autosómica dominante clásica. En la tomografía computarizada y la resonancia magnética, la GCKD aparece como numerosos quistes corticales pequeños. Estos no realzan con gadolinio en la resonancia magnética. A diferencia de otras enfermedades renales quísticas, los túbulos en GCKD generalmente no se ven afectados. Es clínicamente difícil de diagnosticar y, en general, se requiere de la biopsia renal para su diagnóstico (Lundquist AL. Hereditary Renal Cystic Diseases: Glomerulocystic Kidney Disease. In: Renal & Urology News: www.renalandurologynews.com... Accessed February 26th, 2019 [Link to the website]).
Quistes glomerulares en asociación con quistes tubulares se pueden encontrar en varios síndromes que incluyen el complejo de esclerosis tuberosa, el síndrome orofaciodigital 1 y el síndrome de Meckel-Gruber. Los quistes glomerulares también se pueden encontrar en pacientes con mutaciones en los genes UMOD y HNF1β, y en pacientes con enfermedad renal poliquística autosómica dominante con mutaciones PKD1, otras ciliopatías como nefronoptisis, riñón displásico multiquístico u obstrucción del tracto urinario. Sin embargo, la base genética de la enfermedad renal glomerulocística primaria sin dilatación tubular sigue siendo en gran parte desconocida (Rasouly HM, et al. Loss of Zeb2 in mesenchyme-derived nephrons causes primary glomerulocystic disease. Kidney Int. 2016;90(6):1262-1273. [PubMed link]).
Si hay evidencia de alguna enfermedad renal asociada con quistes glomerulares (como displasia renal o esclerosis tuberosa) no debería diagnosticarse como otra enfermedad diferente,
Ver el Caso 157 de nuestra serie de casos: Enfermedad renal glomeruloquística.
ENFERMEDAD QUÍSTICA LOCALIZADA
Es una alteración poco frecuente y de etiología desconocida. Se ha descrito como una alteración multiquística unilateral afectando sólo parte del riñón. Las características morfológicas son similares a las de la EPRAD. Se cree que algunos casos descritos como EPRAD con compromiso unilateral, podrían corresponder a esta enfermedad.
Son quistes encontrados fuera del contexto de una enfermedad poliquística o de una insuficiencia renal crónica. Suelen encontrarse incidentalmente y ser asintomáticos. Su incidencia aumenta con la edad y se encuentran en más de la mitad de personas mayores de 50 años. Son más frecuentes en pacientes con arterioesclerosis y aquellos con fibrosis intersticial.
Su etiología no es clara pero podrían relacionarse con divertículos en túbulos renales. Pueden ser únicos o múltiples, uni o bilaterales y encontrarse en corteza o médula. No conectan con la pelvis o con los cálices. Son esféricos u ovoides y muchos se evidencian bajo la cápsula renal. La pared es lisa, tanto en su parte externa si están bajo la cápsula renal, como en su revestimiento interno; contienen líquido amarillo, a veces turbio y, ocasionalmente, con hemorragia. El tamaño es variable, desde milímetros hasta más de 10 cm. Son uniloculados, pero, ocasionalmente tienen septos internos o lobulaciones. Microscópicamente están tapizados por un epitelio simple aplanado que puede ser discontinuo; su pared está formada por tejido fibroso.
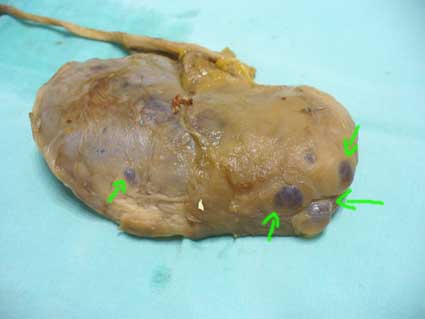
Figura 5. Los quistes simples se ven, con frecuencia, subcapsulares, con pared translúcida y contenido claro amarilloso. Son muy frecuentes en pacientes mayores y usualmente pequeños, sin embargo, podemos ver casos mayores de 5 cm. Con frecuencia son múltiples (flechas). Este riñón corresponde al de un hombre de 64 años fallecido por linfoma sistémico; tenia HTA de larga evolución y su creatinina la última semana antes de su muerte fue de 1,2 mg/dL.
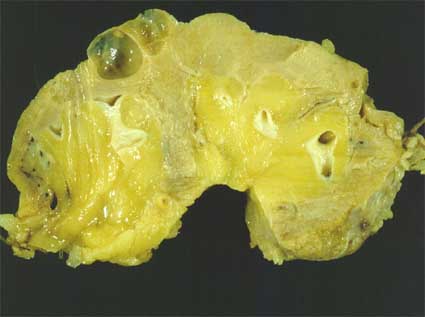
Figura 6. Un par de quistes simples en el riñón de un hombre de 62 años con insuficiencia renal crónica y un carcinoma convencional en otra área del mismo riñón (no mostrado en la fotografía).
Ocasionalmente un quiste puede infectarse o presentar hemorragia. Si está cerca a la pelvis y es de gran tamaño, puede producir obstrucción. Su principal problema radica en que pueden confundirse con neoplasias; por fortuna, usando las técnicas de imagen actuales, es poco probable. Algunos autores han asociado quistes renales simples y neoplasias, pero parece poco probable que exista una relación causal.
También llamada riñón de diálisis. Este diagnóstico está restringido a la enfermedad multiquística asociada con falla renal terminal (usualmente en diálisis crónica) y no causada por una enfermedad renal quística primaria.
Puede presentarse en pacientes con daño renal crónico que no están en diálisis, pero lo usual es que estén en hemodiálisis o en diálisis peritoneal. Su incidencia incrementa con la duración del tiempo en terapia dialítica. Se encuentran quistes renales en aproximadamente el 80% de pacientes en diálisis por más de 4 años. Por definición se requieren al menos 5 quistes por riñón. Algunas series informan más alta incidencia en afroamericanos. Su etiología es desconocida.
La mayoría de casos son asintomáticos; puede haber hemorragia retroperitoneal e infecciones que causan dolor, y, ocasionalmente, pueden tener gran tamaño como para causar dolor abdominal.
Macroscópicamente tienen un tamaño variable; los riñones presentan los cambios crónicos de su daño irreversible; el aspecto de los quistes suele ser similar al de los quistes simples, pero hay un número elevado, a veces cientos. Microscópicamente están tapizados por epitelio simple plano o cúbico; algunas veces con proyecciones papilares. En algunos quistes podemos encontrar un epitelio de células columnares o un epitelio estratificado, algunas veces, incluso, con atipia.
Se asocian con un incremento en la incidencia de carcinoma renal que parece ser proporcional al tiempo en diálisis y mayor en el sexo masculino. Los tipos más frecuentes de carcinoma en estos pacientes son el papilar y el convencional, y algunas veces son múltiples.
También conocido como nefroma quístico (o muliquístico), nefroma mesoblástico quístico, nefroblastoma poliquístico y nefroblastoma diferenciado quístico.
Son lesiones raras que se presentan con dos
picos de incidencia: en menores de 2 años de edad y hacia la edad media
adulta. Se presentan más frecuentemente en polos inferiores, unilateral
y no asociado a enfermedad familiar. En menores de 5 años se ha planteado
que puede ser un tumor de Willms completamente diferenciado. En adultos suele
presentarse entre los 40 y 69 años. 80% de las pacientes con esta lesión
son mujeres. Es una lesión completamente Benigna.
Macroscópicamente la lesión es bien definida, encapsulada, confinada al riñón, de tamaño variable y puede abultar hacia la superficie externa del órgano. El riñón adyacente es normal, salvo por la compresión. Al corte se identifica un cápsula fibrosa que proyecta septos que la dividen en múltiples cavidades de tamaño variable, con contenido líquido claro. [Imagen macro (link)]
Microscópicamente las cavidades están tapizadas por un epitelio cúbico o aplanado, sin atipia. Se ha dicho que estas células tienen características del epitelio de conductos colectores. Los septos entre los quistes están formados por tejido fibroso vascularizado y, a veces, otros componentes mesenquimales como músculo y cartílago. En los quistes no debe haber acúmulos de céluals claras ni en sus septos, si es así debemos pensar que se trata de un carcinoma renal multiquístico de células claras. Tampoco debemos encontrar estroma renal en los septos que separan los quistes, si es así debemos pensar en alguna enfermedad multiquística. Tampoco debemos encontrar elementos embrionarios o blastema (posible tumor de Wilms). [Imagen micro (link)] [Otra imagen micro (link)]
Se ha sugerido que los quistes multiloculares constituyen el "extremo benigno" del continuum de tumores originados del blastema metanéfrico, con el "extremo maligno" representado por el tumor de Wilms. Este concepto está basado en la existencia de "lesiones intermedias" entre las dos: nefroblastoma diferenciado parcialmente quístico.
De todas formas recordemos que es una lesión completamente benigna, no metastatizante.
El riñón, al igual que otros órganos, puede ser afectado por malformaciones que incluyen agenesia unilateral (aproximadamente 1 en 5.000 nacimientos); agenesia bilateral (alrededor de 1 en 7.000 nacimientos); adisplasia renal: entendida como agenesia de un riñón y displasia en el contralateral; hipoplasia renal, ectopias, malrotaciones, fusiones, riñones supernumerarios, duplicación renal etcétera. Aqui sólo mencionaremos algunas por su mayor frecuencia y relevancia clínica.
El término designa un grupo de condiciones caracterizadas por diferenciación y desarrollo anormal del riñón. El término "displasia" no tiene aquí el mismo significado que en otras áreas de la patología general, no designa ningún tipo de atipia celular o condiciones premalignas.
La displasia renal tiene una presentación clínica y aspectos morfológicos muy variables dependiendo de la extensión de la lesión, del compromiso unilateral o bilateral, de si se acompañan de otras alteraciones urinarias y de la presencia o no de alteraciones quísticas. De acuerdo con su morfología se ha dividido en muchos subtipos: displasia multiquística, displasia hipoplásica, displasia quística difusa, displasia aplásica, displasia en sistema duplicado (con sistema pielocalicial y ureter duplicados), displasia asociada con obstrucción o con reflujo, etcétera. Su causa es desconocida.
Se encuentra con frecuencia en mortinatos y recién nacidos con otras malformaciones. Macroscópicamente puede ser uni o bilateral. Los riñones displásicos son pequeños y de forma irregular, si hay marcada hipoplasia puedenser difíciles de identificar. En algunos casos hay quistes, si son múltiples la denominamos displasia renal multiquística. El compromiso puede ser también de sólo una porción del riñón. En ocasiones identificamos islas de cartílago.
Microscópicamente se caracteriza por la presencia de tejido renal inmaduro, con conductos primitivos de células cilíndricas, rodeados por un estroma laxo de aspecto diferente al del riñón normal, con características fetales o embrionarias. Si hay glomérulos tienen un aspecto poco desarrollado y son pequeños. Podemos identificar, con mucha frecuencia, cartílago en medio del tejido displásico.
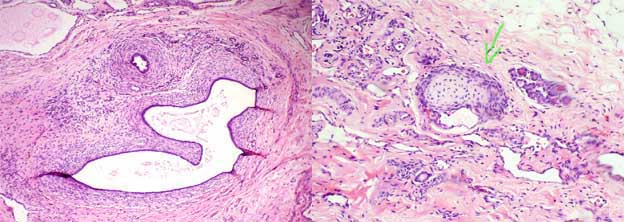
Figura 7. A la izquierda podemos ver un estroma celular primitivo (embrionario o fetal) que rodea conductos dilatados con epitelio cilíndrico o cúbuco que recuerda el tejido renal inmaduro. A la derecha se observa una isla de cartílago hialino, muy frecuente en la displasia renal. (H&E, X300).
El pronóstico depende de la extensión del compromiso renal, de las alteraciones asociadas en vías uriarias y de otras malformaciones presentes.
[Imagen micro (link)] [Displasia renal multiquística varias imágenes macro y micro, click a la flecha (link)]
Puede deberse a agenesia o hipoplasia contralateral (hipertrofia compensatoria). El aumento congénito de la masa renal se encuentra principalmente en síndrome de Wiedemann-Beckwith, hipoglicemia hiperinsulinémica y en el síndrome de Perlman. En estos casos hay aumento del tamaño renal, sin otras alteraciones específicas. En los síndromes anteriores (no en la hiperinsulinemia) puede haber múltiples nidos nefroblastematosos e incremento de la incidencia de nefroblastoma (tumor de Wilms).
OLIGOMEGANEFRONIA (HIPOPLASIA OLIGONEFRÓNICA)
Es una hipoplasia renal bilateral no familiar en la que hay riñones disminuidos de tamaño (<50%) y microscópicamente hay reducción del número de nefronas, los glomérulos están aumentados de tamaño, hasta el triple del normal, y los túbulos están dilatados y con células grandes. Hay poliuria, polidipsia y defectos de concentración de la orina. Son comunes la deshidratación, los vómitos y el retraso en el crecimiento. Hay proteinuria significativa (contrario a la nefronoptisis familiar). Hay evolución a falla renal hacia la segunda década de la vida.
Ver caso 137 de nuestra serie de casos: Oligomeganefronia (con una discusión más amplia de la enfermedad).
Una condición caracterizada por oligohidramnios, suturas craneana muy separadas, parto prematuro y muerte neonatal; puede presentarse el síndrome de Potter por el oligohidramnios e hipoplasia pulmonar secundaria. Parece ser una enfermedad hereditaria. Microscópicamente los glomérulos se ven muy agrupados y los túbulos aparecen con células pequeñas, de tinción oscura y no pueden diferenciarse sus porciones. Por inmunohistoquímica muestran características de túbulos distales. Al parecer hay disminución de la perfusión que lleva a isquemia cortical. Es una afección poco frecuente y, como tantas otras, de causa desconocida.
RIÑÓN EN HERRADURA (FUSIÓN RENAL)
Hay unión del parénquima renal de ambos riñones en la línea media y continuidad entre ellos, usualmente en el polo inferior. Se presenta en aproximadamente 1 de cada 600 personas. En muchos casos es asintomática pero puede asociarse con otras malformaciones de vías urinarias o producir compresión de ureter o de arterias renales.
Principales fuentes bibliográficas:
Bibliografía reciente