
Nefropatía IgA y Púrpura
de Schönlein-Henoch
Patogénesis - Clínica - Histopatología - Púrpura de Schönlein-Henoch
La nefropatía IgA (NIgA) se define por depósitos difusos predominantemente de inmunoglobulina A (IgA) en el mesangio glomerular. El aspecto histológico es muy variable, siendo más frecuentes las alteración mesangiales: proliferación celular y/o de la matriz. Clínicamente se suele presentar con hematuria micro persistente o macro episódica, y proteinuria moderada. Aunque en la literatura médica de antes de los años sesenta hay descripciones de alteraciones que podrían corresponder a NIgA, es sólo con los trabajos de Berger y Hinglais en 1968 y 1969 que la enfermdedad ha sido bien reconocida y clasificada. Se le conoce también como enfermedad de Berger, glomerulonefritis mesangial IgA y enfermedad mesangial IgA.
La NIgA puede ser primaria o secundaria a otras enfermedades, principalmente enfermedad hepatobiliar de cualquier causa. Otras enfermedades asociadas con NIgA son: enfermedad celíaca, enfermedad de Crohn, colitis ulcerativa, síndrome de Sjögren, enfermedades infecciosas o inflamatorias crónicas de mucosas (especialmente de pulmón), enfermedades autoinmunes como espondilitis anquilosante y dermatitis herpetiforme, enfermedades linfoproliferativas, carcinomas de colon, estómago, mama y pulmón.
La púrpura de Schönlein-Henoch, también conocida como síndrome de S-H, nefritis de S-H, púrpura anafilactoide y púrpura reumatoidea (en algunos textos el epónimo aparece invertido: Henoch-Schönlein), está caracterizada por lesiones purpúricas en la piel (principalmente extremidades inferiores y regiones glúteas), artralgias migratorias, hemorragia gastrointestinal y afectación renal. El síndrome, descrito por Schönlein en la primera mitad del siglo XIX, y Henoch, en la segunda mitad del mismo siglo, se considera una forma de NIgA con prominente compromiso extrarrenal: vasculitis sistémica mediada por complejos inmunes ricos en IgA (ver más adelante).
La NIgA es reconocida como la glomerulonefritis primaria más frecuente en todo el mundo y es la causa de insuficiencia renal terminal en aproximadamente el 10-15% de todos los pacientes que llegan a este estado. La enfermedad es más común en el sur de Europa, Asia y entre nativos americanos. Es diagnosticada en el 5-44% de todas las biopsias de acuerdo a diferentes series: 5-10% en Norteamérica y Reino Unido, 20-35% en algunas series europeas y 25-45% en Asia. Las políticas o protocolos con respecto a la toma de biopsias renales responden por parte de esta variabilidad; en centros donde no suelen biopsiarse casos de hematuria microscópica aislada la incidencia será menor; de igual manera, en lugares donde se hace control con uroanálisis, en asintomáticos, para detectar hematuria, habrá mayor porcentaje de casos. Algunos trabajos sugieren que la prevalencia de NIgA subclínica es mucho más alta de lo que parece.
La NIgA puede presentarse a cualquier edad, pero, aproximadamente el 65% de pacientes están en su segunda o tercera décadas de la vida; la edad media del diagnóstico es a los 28 años; menos del 15% de pacientes están por encima de los 40 años de edad y no es muy común en menores de 10 años. La relación hombre:mujer es de aproximadamente 2:1, pero en series de Asia la relación es cercana a 1:1 y en Norteamérica y Europa es de 3,1:1. Afecta todas las razas, habiendo una incidencia más alta en nativos americanos. En algunas series parece haber menos incidencia en africanos y afroamericanos, sin embargo, este dato podría deberse a menor acceso a atención médica y mayor incidencia de otras enfermedades renales más severas (Sehic AM et al, Pediatr Nephrol 11:435-7, 1997 [PubMed link]).
El desarrollo de la NIgA parece requerir un autoanticuerpo (IgG o IgA) contra el autoantígeno IgA1 deficiente de galactosa (Wyatt RJ, Julian BA. IgA nephropathy. N Engl J Med. 2013;368(25):2402-14). Desde el punto de vista anatomopatológico, la NIgA es una glomerulonefritis mediada por complejos inmunes con IgA dominante o codominante, que puede tener una codeposición de IgG por inmunofluorescencia (IF). Sin embargo, como en muchas otras glomerulonefritis, la causa de NIgA no está clara, aunque conocemos algunos aspectos patogénicos. Los depósitos mesangiales son predominantemente de IgA polimérica, subclase 1 (pIgA1). La evidencia actual sugiere que la nefropatía por IgA no se debe a un solo insulto patogénico, sino más bien al resultado de múltiples "golpes" patogénicos secuenciales. Un nivel anormalmente aumentado de IgA1 pobremente O-galactosilada circulante y la producción de anticuerpos específicos de O-glicano conduce a la formación de complejos inmunes que contienen IgA1, y su posterior depósito mesangial da como resultado inflamación y lesión glomerular. Si bien este marco general ha formado la base de nuestra comprensión actual de la patogénesis de la nefropatía por IgA, se está trabajando mucho para tratar de definir con precisión la base genética, epigenética, inmunológica y molecular de la nefropatía por IgA. En particular, se sigue evaluando intensamente el origen preciso de la IgA1 pobremente O-galactosilada y los factores que incitan a la producción de anticuerpos específicos de O-glicano. Los mecanismos responsables de la deposición de IgA1 mesangial y la lesión renal posterior tampoco están bien entendidos. (Yeo SC, et al. New insights into the pathogenesis of IgA nephropathy. Pediatr Nephrol. 2018;33(5):763-777).
Tres elementos claves se relacionan con el desarrollo, severidad y evolución de la NIgA (Barratt J y Feehally J, J Am Soc Nephrol 16:2088-97, 2005 PubMed link): 1.) Síntesis, liberación y persistencia en la circulación de pIgA1 con características que favorecen su depósito mesangial; 2.) La "reactividad" del mesangio: susceptibilidad a que se deposite pIgA1 y capacidad de generar respuesta inflamatoria a los depósitos; y 3.) la respuesta del riñón a la agresión y su tendencia a montar una cascada de eventos que favorece mayor lesión (daño túbulo-intersticial y glomeruloesclerosis) y no la resolución de ésta. Cada uno de estos tres elementos puede tener una base genética que influencia el fenotipo de la enfermedad en cada individuo.
Han sido descritas muchas alteraciones en la IgA y en su producción, sin embargo, las alteraciones son heterogéneas en diferentes grupos de individuos. Este concepto respalda la hipótesis de que hay varios mecanismos patogénicos de la enfermedad. Sólo el incremento de la IgA circulante no es suficiente para producir depósitos mesangiales; son necesarias otras características especiales que promueven su localización allí.
No se ha demostrado que la IgA sea un autoanticuerpo contra antígenos mesangiales. Por el contrario, en muchos casos se ha documentado que la IgA circulante en estos pacientes está formada por anticuerpos IgA1 contra diferentes antígenos, sin identificarse uno en particular como patogénico. De otro lado, se han encontrado varias alteraciones inusuales en las características físicas de la molécula, sin saberse cuál de éstas es responsable de su depósito en el mesangio y de la activación de células mesangiales. Una de las alteraciones estructurales más estudiada es la glicosilación anormal de moléculas de IgA1. Esta alteración generaría una tendencia de las moléculas a auto-agregarse y formar complejos antígeno-anticuerpo con anticuerpos IgG dirigidos contra epítopes de IgA1, favoreciendo la formación de agregados macromoleculares de pIgA1 y complejos inmunes IgA que promueven su depósito mesangial. Además, este tipo de moléculas IgA1 alteradas tienen más afinidad, in vitro, por componentes de la matriz extracelular: fibronectina y colágeno tipo IV.
El sitio de producción de la IgA en NIgA ha sido objeto de múltiples estudios. La asociación entre episodios de hematuria macro e infecciones de las mucosas llevó a sospechar la presencia de antígenos anormales en la mucosa, debido principalmente a que ambas, la IgA sérica y la IgA depositada en el mesangio, son poliméricas. Sin embargo, estudios posteriores han demostrado que la sobreproducción de pIgA no puede explicarse sólo por sobreproducción en mucosas y salida a la circulación, estos estudios demuestran un número normal o reducido de células plasmáticas productoras de IgA en la mucosa y no hay incremento de pIgA en las secresiones mucosas o, incluso, puede estar reducida. En cambio, sí se ha documentado incremento de células plasmáticas productoras de pIgA en la médula ósea. Por lo tanto la sobreproducción sería un evento sistémico, probablemente originado como respuesta a antígenos en mucosas (Barratt J y Feehally J, J Am Soc Nephrol 16:2088-97, 2005 [PubMed link]). Reforzando este concepto, sabemos que la IgA producida en mucosas es predominantemente IgA2 y la que se deposita en glomérulos IgA1.
También se ha propuesto una alteración en la eliminación de IgA y de complejos inmunes IgA, lo cual facilitaría su persistencia en la circulación. Uno de los mecanismos de degradación de la IgA es por un receptor Fc: CD89, el cual media su endocitosis y catabolismo. En NIgA hay una subregulación del CD89 en células mieloides lo que llevaría a disminución en su eliminación. También se a demostrado menor afinidad de IgA por el receptor CD89 en NIgA.
No todos los depósitos de IgA generan inflamación mesangial ni estos depósitos son irreversibles. Es probable que el incremento de macromoléculas de IgA facilite su atrapamiento inespecífico en el mesangio, quizá incrementado por el defecto en la glicosilación de la molécula. También se ha sugerido la formación de complejos CD89-IgA libres en la circulación que se depositarían en el mesangio.
Hay evidencia in vitro de que las células mesangiales pueden captar y catabolizar moléculas de IgA mediado por receptores y endocitosis. Es posible que alguna alteración en el mecanismo de degradación lleve a acumulación mesangial.
La NIgA no está, en general, asociada con migración de células inflamatoria al mesangio, sugiriendo que gran parte de la lesión se debe a expansión de las células glomerulares residentes. La lesión glomerular ocurre predominantemente através de activación del complemento inducida por IgA. La exposición a IgA sobrerregula la síntesis de matriz extracelular y un estado proinflamatorio y profibrótico de las células mesangiales. Aunque la activación del complemento no es esencial para el desarrollo de NIgA, hay evidencia que sugiere que su activación puede influir en la extensión del daño glomerular. La activación de C3 por IgA mesangial lleva a la generación de C5b-C9 que activaría la célula mesangial generando producción de mediadores de la inflamación y de componentes de la matriz extracelular. Al igual que en nefritis lúpica, el C3 depositado en el mesangio se encuentra en forma activada, enfatizando la importancia de la activación del complemento en esta enfermedad.
Hay factores genéticos relacionados con el desarrollo de la enfermedad y es bien conocida la existencia de formas familiares. Sin embargo, hasta ahora no hay genes candidatos. Estudios en relación con diferentes genes y loci nos darán alguna luz en un futuro, ojalá cercano.
La NIgA comunmente recurre en aloinjertos, aunque raras veces es clínicamente significativa, y en riñones de donantes con depósitosa de IgA éstos desaparecen post-trasplante; estos datos indican algún factor extrarrenal subyacente a la nefropatía.
NIgA secundaria: en el caso de NIgA secundaria a enfermedad hepática, parece importante el papel que desempeña el hígado en la depuración de moléculas de IgA, principalmente de IgA1, los hepatocitos son la mayor ruta de degradación de IgA. Además del incremento en los niveles séricos de IgA, otros mecanismos juegan un papel importante en la patogénesis de la NIgA secundaria a enfermedad hepatobiliar: Alteración en la función de la fagocitosis en monocitos, acumulación de otras sustancias nocivas, mayor riesgo de infección, hipocomplementemia e incremento en la producción de TGF-beta.
En la NIgA asociada a enfermedades de las mucosas parece jugar un gran papel la sobreproducción de IgA, sin embargo, otros mecanismos, poco entendidos, deben ser importantes en la patogénesis; quizá producción de algunas citoquinas o alteraciones de la barrera mucosa al paso de antígenos. En la NIgA secundaria a desórdenes de la inmunidad y enfermedades linfoproliferativas, la patogenia es muy poco clara; desorden en la producción de factores de crecimiento, alteraciones de células T, producción alterada de citoquinas, etcétera, han sido mecanismos relacionados con la patogénesis.
Clínica: Hematuria es la manifestación clínica más consistentemente expresada en NIgA, es observada en algún momento de la evolución en más del 95% de pacientes. La microhematuria persistente ocurre en aproximadamente el 78% de casos y macrohematuria episódica en 54%. La macrohematuria suele presentarse por unos pocos días y se detecta microhematuria entre estos episodios. En otros casos puede haber microhematuria intermitente. En algunas series la macrohematuria es más frecuente en niños, mientras otras series encuentran poca diferencia entre adultos y niños. Otro signo frecuente es la proteinuria, usualmente <1g/24h, aunque un 5-10% tendrán proteinuria severa, o aun desarrollarán síndrome nefrótico completo. Hay hipertensión en un 15% de pacientes y elevación de creatinina en 10%. En 5% de casos se detectara falla renal crónica al momento del diagnóstico. Es menos probable encontrar HTA y falla renal en niños. En unos pocos pacientes hay dolor abdominal o en flancos.
En muchos casos en los que la NIgA se presenta como macrohematuria episódica, los cuadros se presentan asociados a faringitis; a diferencia de la GN postestreptococcica, en la NIgA los episodios de hematuria suceden con o poco después de la faringítis: sinfaringítica; la separación entre el inicio de la faringitis y la hematuria es de días, más que de semanas como en la GN postinfecciosa (postfaringítica). Episodios de gastroenteritis, sinusitis o bronquitis están también asociados con la hematuria. A pesar de haber, al menos en algunos casos, lesiones histológicas de mayor grado en los episodios de macrohematuria, algunos trabajos muestran mejor pronóstico para los pacientes con esta manifestación clínica.
En algunos pacientes hay insuficiencia renal aguda asociada a episodios de hematuria; esta falla renal suele ser transitoria y no parece afectar el pronóstico. En muchos casos está asociada a daño tubular debido a hemoglobina. En otros casos puede haber GN extracapilar asociada a episodios de macrohematuria y GN rápidamente progresiva, con un peor pronóstico: mayor evolución a daño renal crónico. En los pacientes con microhematuria persistente o episódica hay menos incidencia de falla renal aguda, pero mayor probabilidad de disminución progresiva de la tasa de filtración glomerular.
En pacientes con NIgA y síndrome nefrótico se ha propuesto que hay una colisión entre NIgA y glomerulopatía de cambios mínimos (CGM), sin embargo, aunque suele ser imposible en este contexto comprobar dicha coexistencia de enfermedades, el número de tales pacientes excede el esperado según la frecuencia de CGM. Más fácil de comprobar ha sido la colisión de NIgA y GN membranosa, aunque es un evento inusual. El síndrome nefrótico en el contexto de una NIgA podría ser el resultado de un proceso fisiopatogénicamente diferente.
Aunque inicialmente considerada una enfermedad benigna, sabemos que la NIgA puede llevar a falla renal terminal en aproximadamente el 30% de casos a los 20 años del diagnóstico. En muchos casos hay remisión espontánea, pero es sostenida en sólo un 10% de casos. Entre el 60% y el 70% continuará con episodios de exacerbación y relativa quietud de la enfermedad. La NIgA es la causa de enfermedad renal terminal en alrededor del 10-15% de todos los casos.
NIgA recurre post-trasplante en aproximadamente el 50% de casos: manifestada como depósitos de IgA, sin embargo, el riesgo de disfunción significativa es del 13% y el de falla renal de 5% a cinco años.
No hay una terapia efectiva y bien establecida para la enfermedad. Es importante el control de condiciones subyacentes como la HTA. Los esteroides parecen de poca utilidad, aunque en pacientes en los que hay síndrome nefrótico se informa una mayor proporción de respuesta. Agentes bloqueadores de los canales del calcio e inhibidores de la enzima convertidora de angiotensina parecen tener alguna utilidad aun en pacientes sin HTA. También ha ganado aceptación el uso de aceites ricos en ácidos grasos omega-3, sin embargo pocos trabajos demuestran su eficacia y otros no encuentran beneficio. Inmunosupresores y otros tratamientos han sido usados con resultados variables. En algunos casos se recomienda amigdalectomía. En muchos pacientes no se da ningún tratamiento, sólo se hace vigilancia y control periódicos.
Datos de laboratorio: Hay hematuria, en algún momento de la evolución, en el 95% de casos. La proteinuria es muy variable, pero lo más habitual es que sea <1g/24h, la creatinina sérica y el BUN están aumentados en unos pocos pacientes al momento del diagnóstico. Los niveles séricos de IgA pueden estar incrementados en un buen número de pacientes, pero no constituyen una prueba con alta sensibilidad ni especificidad para el diagnóstico. No hay alteraciones en los niveles del complemento.
El aspecto histopatológico de la glomerulopatía es muy variable, desde glomérulos con aspecto normal hasta cualquier tipo de lesión proliferativa o esclerosante, incluyendo proliferación extracapilar. Las lesiones predominantes son la proliferación mesangial (de células y matriz) y la nefritis proliferativa focal; NIgA es la primera posibilidad cuando se presenta este cuadro histológico en un paciente sin enfermedad sistémica. La glomerulopatía mesangiopática es la presentación morfológica en más del 60% de casos; en general hay incremento de células y matriz mesangial en grados variables. La alteración tiende a ser más o menos uniforme en todos los glomérulos, pero en algunos casos hay gran variación entre ellos en una misma biopsia. La expansión mesangial es más notoria con el PAS que con la plata-metenamina debido a las características de los depósitos mesangiales; el grado de esta expansión es difícil de determinar con precisión, es algo subjetivo y con poca reproducibilidad entre observadores, a menos que se haga por análisis morfométrico. Los depósitos inmunes no suelen verse con microscopía de luz convencional.
Figura 1. Penacho glomerular con leve hipercelularidad mesangial global; la inmunofluorescencia directa demostró depósitos mesangiales difusos de IgA. Mujer de 29 años con hematuria recurrente macro y proteinuria leve, la función renal no presentaba alteraciones (H&E, X400).
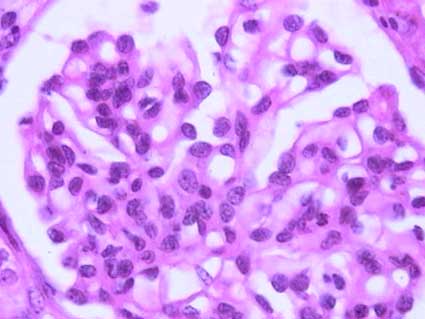
Figura 2. La hipercelularidad mesangial se ve más claramente en este glomérulo de un paciente de 45 años con nefropatía IgA. (H&E, X400).
En algunos casos las lesiones mesangiales son focales y segmentarias. En otros hay proliferación endocapilar focal y segmentaria, con infiltración de leucocitos (aproximadamente 17% de casos) y en casi un 5% las lesiones proliferativas endocapilares son difusas. En una proporción variable de casos se identifican lesiones eclerosantes segmentarias y focales; puede haber sinequias a la cápsula de Bowman y segmentos con hipertrofia e hiperplasia de podocitos. En unos pocos casos hay segmentos de necrosis del penacho glomerular. Podemos ver semilunas en glomérulos con cualquiera de los tipos de lesión del penacho; en 5-8% de pacientes las semilunas comprometen más del 50% de glomérulos. Se encuentran depósitos subepiteliales frecuentemente asociados a las semilunas. También podemos ver otros patrones de lesión glomerular en NIgA: GN membranoproliferativa, membranosa y lesiones glomerulonefríticas mixtas. Alrededor de un 12% de casos muestran glomérulos normales o mínimamente alterados.
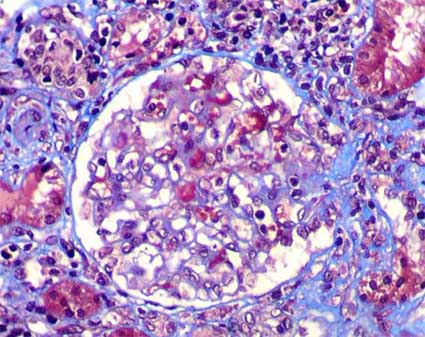
Figura 3. En algunos casos de nefropatía IgA encontraremos lesiones glomerulares esclerosante segmentarias como en esta biopsia de un hombre de 37 años con hematuria micro persistente. Estas lesiones pueden indicar un grado mayor de severidad de las alteraciones renales, pero, no necesariamente se correlacionan con la proteinuria o con la función renal. (Tricrómico de Masson, X400).
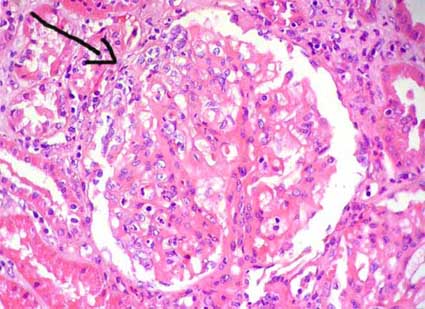
Figura 4. En este glomérulo además de hipercelularidad mesangial hay segmentos con hipercelularidad endocapilar, disminución de luces capilares y células inflamatorias. La flecha señala una pequeña semiluna epitelial circunscrita. (H&E, X400).
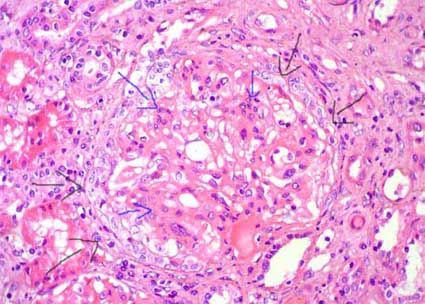
Figura 5. Mismo caso de la microfotografía anterior; observe el penacho comprimido por una semiluna epitelial circunferencial (flechas negras); hay proliferación mesangial celular y de la matriz (flechas azules). Este caso corresponde al de una mujer de 32 años con episodios de hematuria macro, en el último, previo a la bopisia, presentó además elevación de la creatinina. Las semilunas extensas en NIgA se asocian con un curso clínico más agresivo. (H&E, X400).
En los túbulos pueden identificarse eritrocitos o cilindros eritrocitarios, pero su ausencia no implica que el paciente no ha tenido hematuria. Hay cambios de daño epitelial tubular en hasta el 57% de biopsias: Descamación celular, pérdida del borde en cepillo, vacuolización citoplasmática y dilatación tubular; estos cambios se evidencian principalmente en pacientes con falla renal aguda. También pueden evidenciarse otros signos de daño crónico: Atrofia tubular y fibrosis intersticial. En un 5% de casos hay franca inflamación tubulointersticial aguda. Hay fibrosis intimal de arterias, hipertrofia medial y depósitos arteriolares hialinos en una proporción variable de casos.
Han habido varios sistemas para clasificar las lesiones microscópicas de la NIgA, sin embargo, hasta el momento no hay una clasificación universalmente aceptada; diferentes trabajos muestran correlación variable con el pronóstico. Algunas de las más usadas son la de S.R. Meadow (Meadow SR, et al, Q J Med 41:241-58, 1972[PubMed link]), la de M. Haas (Haas M, Am J Kidney Dis 29:829-42, 1997[PubMed link]), la de H.S. Lee (Lee HS, et al, Nephrol Dial Transplant 20:342-8, 2005 [PubMed link]) y la de S.M.K. Lee (Lee SM, et al, Hum Pathol 13:414-22, 1982 [PubMed link]). Estas clasificaciones se basan en la severidad y extensión de la proliferación mesangial, el porcentaje de glomérulos con lesiones segmentarias, con semilunas y con esclerosis global, y, en algunos esquemas, la extensión del daño túbulo-intersticial. La International IgA Nephropathy Network ha elaborado una propuesta de clasificación de la NIgA que espera se correlacione mejor con el pronóstico y defina mejor los tipos y grados de lesión, este trabajo fue auspiciado por la Renal Pathology Society y la International Society of Nephrology y aparece en dos artículos: "rationale, clinicopathological correlations, and classification" y "pathology definitions, correlations, and reproducibility".
Clasificación de Oxford
Publicada inicialmente en 2009 y modificada en 2016 (agregando el "C" score) gradúa 5 parametros: hipercelularidad mesangial (M), hipercelularidad endocapilar (E), lesiones glomerulares esclerosantes segmentarias (S), fibrosis intersticial y atrofia tubular (T) y semilunas (C), así:
Clasificación de Oxford de la nefropatía IgA (MEST-C) (Cattran DC, et al. The Oxford classification of IgA nephropathy: rationale, clinicopathological correlations, and classification. Kidney Int. 2009;76:534-45 [PubMed link]; Trimarchi H, et al. IgAN Classification Working Group of the International IgA Nephropathy Network and the Renal Pathology Society; Conference Participants. Oxford Classification of IgA nephropathy 2016: an update from the IgA Nephropathy Classification Working Group. Kidney Int. 2017;91:1014-1021 [PubMed link]). |
|
M |
M0: No hay hipercelularidad mesangial o ésta compromete <50% de glomérulos M1: >50% de glomérulos tienen >3 células mesangiales en un área mesangial |
E |
E0: Ausente: No hay hipercelularidad endocapilar E1: Presente: Al menos un glomérulo con hipercelularidad endocapilar |
S |
S0: Ausente: No hay lesiones glomerulares esclerosantes segmentarias S1: Presente: Al menos un glomérulo con lesión esclerosante segmentaria |
T |
T0: Fibrosis intersticial o atrofia tubular (IF/TA) en < /= 25% T1: IF/TA en 26-50% T2: IF/TA >50% |
C |
C0: No hay semilunas C1: Semilunas en <25% (celulares o fibrocelulares*) C2: Semiluinas en >/= 25% (celulares o fibrocelulares*) |
* Semilunas fibrosas (compuestas en >/=90% de matriz) no se tienen en cuenta.
Como puede verse, más que una clasificación, es una graduación semicuantitativa de 5 parámetros. Desde un punto de vista personal (opinión) es una "clasificación" que deja de lado mucha información importante, por ejemplo, no es lo mismo si en una biopsia encontramos 1 glomérulo de 40 con esclerosis segmentaria, que otra en la que encontramos 20 de 40 con dicha esclerosis segmentaria, en ambos casos el parámetro "S" es S1, sin embargo, todos sabemos que con tantos glomérulos comprometidos el pronóstico será peor. Esta clasificación tampoco tiene en cuenta el poprcentaje de glomeruloesclerosis global, que, lógicamente, es muy importante como un factor pronóstico. Tampoco tiene en cuenta la presencia de lesiones necrosantes. Más importante que una graduación o clasificación, para definir el manejo de una paciente es esencial analizar todo el contexto clínico y la información detallada de un buen informe del estudio de la biopsia (igual para todas las clasificaciones en patología renal).
Inmunofluorescencia
Los depósitos mesangiales de IgA son el sello característico de la enfermedad, sin embargo, los depósitos son exclusivos de IgA en sólo un 26% de casos; se suelen acompañar de IgG (37%) o IgM (13%), y pueden estar las tres Igs en el 25% de casos. Por definición, la IgA debe ser dominante o codominante; si hay depósitos de IgG o IgM más intensos que los de IgA debemos pensar en otra glomerulopatía. Hay depósitos de C3 en la gran mayoría de casos (95%) y ocasionalmente de fracciones relacionadas con activación de la vía clásica: C1q y C4 (12%). Los depósitos de IgA son mesangiales granulares y difusos en casi todos los pacientes, sin embargo, ocasionalmente son segmentarios. La IgA predominante es la de subclase 1 y en términos de cadenas ligeras predomina la lambda. No hay depósitos del componente secretorio de IgA. En la mayoria de pacientes se detecta el complejo de ataque de membrana (C5b-C9) pero su importancia fisiopatogénica no está muy clara. Algunos estudios han encontrado que la presencia de depósitos de C4d se ha asociado con menor supervivencia renal (Espinosa M, et al. Association of C4d deposition with clinical outcomes in IgA nephropathy. Clin J Am Soc Nephrol. 2014;9(5):897-904 [PubMed Link], Segarra A, et al. Mesangial C4d Deposits in Early IgA Nephropathy. Clin J Am Soc Nephrol. 2018;13(2):258-264 [PubMed link]..
En aproximadamente un 25% de casos hay grados variables de depósitos de IgA en paredes capilares. La codeposición de IgG y la ubicación de los depósitos inmunes glomerulares en las paredes capilares parecen estar asociadas con una mayor actividad histológica en la biopsia renal, pero solo la ubicación de los depósitos inmunes glomerulares en las paredes capilares se asoció con un riesgo significativamente mayor de enfermedad renal terminal, trasplante, muerte y/o duplicación de creatinina sérica (Alvarado AS, et al. Location of glomerular immune deposits, not codeposition of immunoglobulin G, influences definitive renal outcomes in immunoglobulin A nephropathy. Nephrol Dial Transplant. 2018;33(7):1168-1175. [PubMed link]).
Ver Caso 24 de nuestra serie de casos: co-depósitos de IgA e IgM y depósitos subendoteliales.
Ver Caso 153 de nuestra serie: co-depósitos de IgA e IgG y depósitos subendoteliales.

Figura 6. La característica fundamental en la NIgA es la inmunotinción mesangial intensa, difusa, para IgA. Observe que la positividad se limita, en este caso, a los tallos mesangiales, sin comprometer las paredes capilares; esta microfotografía representa el típico patrón mesangial (Inmunofluorescencia con anticuerpos anti-IgA marcados con fluoresceina, X400).
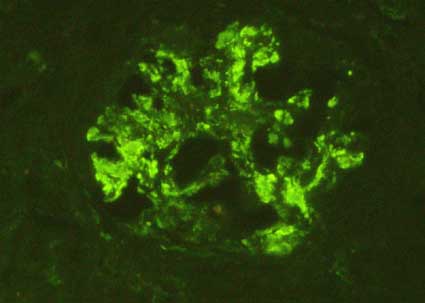
Figura 7. Cuando el ensanchamiento mesangial es marcado, la positividad muestra áreas más amplias dentro del glomérulo. En ocasiones también podemos ver algunos depósitos parietales (en paredes capilares) (Inmunofluorescencia con anticuerpos anti-IgA marcados con fluoresceina, X400).
Microscopía electrónica
Hay depósitos mesangiales electrón-densos que correlacionan con los depósitos inmunes. Muchos de éstos se localizan en la interfase entre el mesangio y las luces capilares, o la unión del mesangio y la MBG: paramesangio. Estos depósitos son de tamaño variable; algunos autores creen que los depósitos ubicados más profundos en el mesangio reflejan estadios más avanzados de lesión. Hay depósitos parietales subendoteliales en aproximadamente un 19% de casos, subepitelailes en 12% e intramembranosos en 13% de los casos. Las células mesangiales presentan incremento de mitocondrias y retículo endoplásmico rugoso.
En algunos casos se detecta laminación y fragmentación de la MBG similar a los cambios vistos en Enfermedad de Alport y en otros trabajos se ha documentado la presencia de membrana basal delgada. La coexistencia de estas lesiones tiene significado clínico y pronóstico aún incierto; no está muy clara su patogénesis. Podría tratarse de una colisión de dos enfermedades glomerulares.
Indicadores pronósticos
Características clínicas asociadas con mayor probabilidad de daño renal crónico incluyen: Edad mayor de inicio de la enfermedad, no presentar episodios de macrohematuria, proteinuria severa (en algunos estudios >1g/24h), hipertensión, disminución persistente de la tasa de filtración glomerular, cretinina sérica >1,4 mg/dL y alteración en la capacidad de concentración de la orina.
Alteraciones histológicas relacionadas con peor pronóstico: Extensión de la fibrosis intersticial y atrofia tubular, glomeruloesclerosis (global o segmentaria), hipercelularidad mesangial, proliferación endocapilar, semilunas, segmentos necrosantes, esclerosis vascular, depósitos inmunes parietales (subendoteliales) y abundantes codepósitos de IgG o de IgM.
Aunque muchas de estas variables han demostrado correlación con el pronóstico en estudios de poblaciones de pacientes, no tienen mucho valor cuando se analiza un caso individual.
Ver "Caso 3" como un ejemplo de nefropatía IgA.
Es una enfermedad caracterizada por nefritis aguda asociada a lesiones purpúricas palpables de la piel, principalmente en extremidades inferiores y regiones glúteas, artritis migratoria y hemorragia gastronitestinal. Es también conocida como síndrome de S-H, nefritis de S-H, púrpura anafilactoide y púrpura reumatoidea (en muchos textos el epónimo aparece invertido: Henoch-Schönlein). El nombre se debe a las médicos alemanes Johann Lukas Schönlein (1793-1864) y Eduard Heinrich Henoch (1820-1910). Algunos autores consideran que el nombre "Schönlein" debería ir en primer lugar debido a que fue la primera persona en reconocer la enfermedad (en 1837) (o al menos eso se cree, ya que la condición parece haber sido primero descrita por Heberden en 1801 [Jennette JC, et al. Heptinstall's Pathology of the Kidney. 6th ed. Wolters Kluwer Lippincott Williams & Wilkins, Philadelphia, 2007]). Henoch en 1868 reportó el primer caso de un paciente con cólico, diarrea sanguinolenta, artralgias y lesiones cutáneas. Se considera una forma de NIgA con prominente compromiso extrarrenal: vasculitis sistémica mediada por complejos inmunes ricos en IgA. Ocurre principalmente en niños, pero puede afectar pacientes de cualquier edad. Las alteraciones histopatológicas pueden ser similares a las de la NIgA, incluyendo su aspecto inmunopatológico y ultraestructural.
La púrpura de S-H es considerada una enfermedad autoinmune mediada por IgA. Se postula que un antígeno desconocido estimula la producción e incremento de la IgA, y que los complejos inmunes así formados se depositan localmente en muchos sitios del cuerpo activando inflamación y originando vasculitis necrotizante. La etiología de la enfermedad se cree que es multifactorial, con componentes genéticos, medioambientales y antigénicos. Muchos pacientes relatan antecedente de infección de vías respiratorias superiores, o gastrointestinales. También se informan casos de púrpura de S-H después de ingestión de algunos medicamentos y de vacunas.
La presentación clínica incluye un rash característico, poliartritis migratoria, compromiso renal y compromiso gastrointestinal. Después de síntomas prodrómicos se desarrollan las lesiones en piel, dolor abdominal, edema periférico, vómito y artritis, aunque sólo las lesiones en piel se presentan en el 100% de casos, siendo el síntoma de presentación incial en el 50% de pacientes. Las lesiones consisten en máculas eritematosas, pápulas urticariales y placas (púrpura palpable). Estas lesiones cutáneas tienden a aparecer en extremidades inferiores, parte baja del abdomen y regiones glúteas. La erupción usualmente dura un promedio de 3 semanas. Hay síntomas gastrointestinales en hasta el 85% de pacientes, e incluyen dolor abdominal, nauseas y vómito, siendo el síntoma más común el dolor tipo cólico. El compromiso articular se presenta en cerca del 75% de casos y es el síntoma de presentación inicial en el 25%. Suelen afectarse articulaciones de rodilla y tobillos, presentando dolor y edema. Las alteraciones renales se presentan entre el 30 y el 50% de pacientes y pueden persistir hasta 6 meses después de las lesiones cutáneas. El compromiso renal se manifiesta con hematuria leve y/o proteinuria, y en algunos casos oliguria y falla renal. Pueden haber alteraciones renales permanentes en hasta el 20% de pacientes.
Aun cuando la presentación clínica característica permite un diagnóstico relativamente fácil, la confirmación del diagnóstico requiere evidencia de depósitos de IgA en tejidos (piel o riñón). La biopsia renal se reserva para pacientes con diagnóstico difícil o pacientes con afectación renal, para determinar severidad de las lesiones y conocer el estado morfológico del órgano.
La severidad de las lesiones glomerulares es muy variable, pero con frecuencia se encuentra proliferación endocapilar, semilunas y segmentos necrotizantes. Todas estas lesiones severas deben cuantificarse en una biopsia en la que se diagnostique la enfermedad. El amplio espectro de lesiones glomerulares asemeja al de la nefritis lúpica, pudiéndose encontrar un patrón de GN membranoproliferativa. Las lesiones pueden ser variables en el tiempo en un mismo individuo. También hay diversas clasificaciones histopatológicas de la enfermedad, con variable correlación con el pronóstico; lo más importante es hacer una rigurosa cuantificación de semilunas, lesiones proliferativas segmentarias o difusas y áreas segmentarias de necrosis y de esclerosis.
Por inmunofluorescencia y microscopía electrónica los hallazgos pueden ser similares a los de la NIgA, con mayor frecuencia de depósitos subendoteliales y subepiteliales, principalmente los primeros. En algunos casos los depósitos parietales predominan sobre los mesangiales. Al igual que en NIgA la subclase predominante en los depósitos es IgA1.
En las lesiones de piel pueden identificarse depósitosa de IgA similares a los depósitos glomerulares.
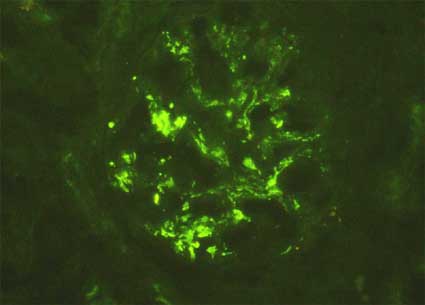
Figura 8. Las carceterísticas inmunopatológicas en el síndrome de Schönlein-Henoch suelen ser similares a las de la NIgA, como en este caso de un paciente de 9 años con la enfermedad; sin embargo, son más frecuentes los depósitos inmunes parietales, principalmente subendoteliales. (Inmunofluorescencia con anticuerpos anti-IgA marcados con fluoresceina, X400).
Las alteraciones renales marcan el pronóstico de la enfermedad. Aproximadamente el 60% de pacientes presenta resolución de la enfermedad, usualmente en los primeros 6 meses; otro 15 a 20% presenta anormalidades urinarias menores sin deterioro de la función renal, y otro 20 a 25% presenta daño renal progresivo hasta falla renal terminal en 10 a 20 años. Hallazgos de mal pronóstico incluyen un curso clínico prolongado, proteinuria severa, hematuria macrocópica, hipertensión arterial, porcentaje de semilunas (mayor del 50%) y, en algunos trabajos, extensión de los depósitos subendoteliales.
En el tratamiento suelen usarse esteroides u otros inmunosupresores, pero no hay acuerdo universal respecto al tratamiento óptimo. La enfermedad puede recurrir post-trasplante (53%), pero usualmente como depósitos de IgA sin deterioro de la función; se detecta nefritis clínica recurrente en aproximadamente 18% de casos; en 9% se producirá pérdida del injerto como consecuencia de la enfermedad.
Ver el Caso 18 y el Caso 45 de nuestra serie de casos.
Bibliografía: