Neoplasias renales
Parte 1. Generalidades - Clasificación - Carcinoma de células renales (CCR) - Estadificación - CCR convencional - Neoplasia renal quística multilocular de bajo potencial maligno - CCR papilar - CCR cromófobo - Carcinoma de conductos colectores - Carcinoma medular - Carcinoma de conductos colectores de bajo grado - Carcinoma tubuloquístico
Parte 2. Carcinoma mucinoso tubular y de células fusiformes - CCr asociado a la translocación Xp11.2 - CCR inclasificable y CCR en enfermedad renal quística adquirida - Adenoma papilar - Oncocitoma - Diagnóstico diferencial en tumores con células granulares - Adenoma metanéfrico - Adenofibroma metanéfrico - Tumor estromal metanéfrico - Angiomiolipoma - Tumor renomedular de células intersticiales
Parte 3. Tumor de células yuxtaglomerulares - Nefroma quístico del adulto y tumor estromal y epitelial mixto (MEST) - Nefroma quístico pediátrico - Sarcoma sinovial - Nefroblastoma (tumor de Wilms) - Nefroblastoma quístico parcialmente diferenciado - Restos nefrogénicos (y nefroblastomatosis) - Sarcoma de células claras - Tumor rabdoide - Nefroma mesoblástico congénito - Tumor renal osificante de la infancia
Parte 3
Tumor de células yuxtaglomerulares (reninoma)
Es un tumor muy poco frecuente, localizado en la corteza, que se origina de las células del aparato yuxtaglomerular y frecuentemente se asocia con hipertensión por exceso de producción de renina. Se presentan en adultos jóvenes y en adolescentes (edad media 23-27 años) y, aunque hay pocos casos en la literatura, parecen ser más frecuentes en mujeres (H:M: 1:2). Son lesiones benignas, es muy inusual la presencia de metástasis o de recurencia. En el diagnóstico la medición de renina plasmática por cateterización selectiva de la vena renal es uno de los metódos más efectivos para reconocer este tipo de tumor.
Macroscópicamente son lesiones usualmente menores de 3 cm, bien circunscritas, de color crema o grisáceos, pueden contener pequeños espacios quísticos. Microscópicamente tienen un aspecto variable: células poligonales con disposición trabecular en un estroma mixoide; podemos ver disposición en nidos o un patrón "organoide" similar al de muchos carcinoides; en otros casos podemos ver islas sólidas, túbulos, papilas y quistes. La vascularización es prominente y puede ser similar a un hemangiopericitoma.
El citoplasma muestra gránulos positivos con el PAS. Por IHQ hay positividad difusa, citoplasmática, para renina. La microscopía electrónica muestra características estructuras electron-densas que pueden ser redondas (gránulos maduros) o romboidales (protogránulos de renina).
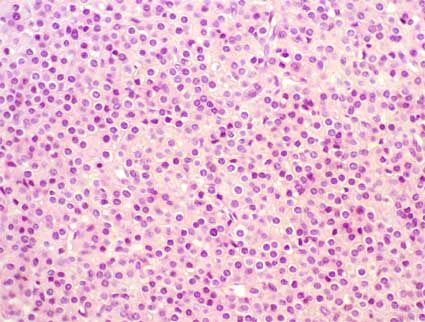
Figura 29. Tumor de céluas yuxtaglomerulares. Hay una disposición sólida de células con limites bien definidos, núcleos centrales, redondos, homogéneos, sin o con muy escasas mitósis. En algunas áreas hay disposición en nidos de las células tumorales, algo similar a carcinoides. (H&E, X400).
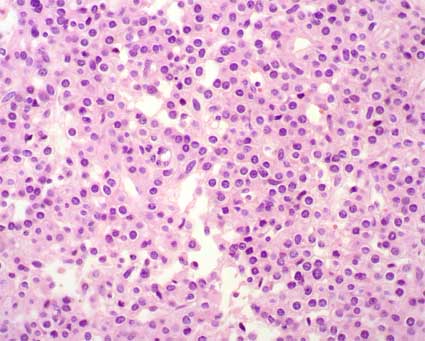
Figura 30. En este caso hay disposición menos densa de las células y un patrón algo hemangiopericitoide. (H&E, X400).

Figura 31. La microscopía electrónica, al igual que la inmunohistoquímica, permiten confirmar la presencia de gránulos (redondos) y protogránulos (romboidales) de renina. Las flechas azules señalan gránulos redondos de renina madura y las verdes protogránulos romboidales de la enzima; ambos son característicos de reninomas. (Microscopía electrónica, X6000. Imagen cortesía de la Dra. Julia Blanco, Hospital Clínico San Carlos, Madrid, España).
Importante: la producción de renina y positividad por IHQ para ésta, no es exclusiva de los reninomas, otros tumores pueden presentar esta característica: carcinomas de células renales, nefroblastoma, adenocarcinomas pancreáticos, hepatoblastoma, carcinoma de pulmón y otros. Además, cualquier tumor que comprima la arteria renal puede llevar a un incremento en la producción de renina.
Tumores epiteliales y estromales mixtos
En la clasificación de tumores de la OMS 2022, este grupo de tumores comprende el tumor estromal y epitelial mixto (MEST por su sigla en inglés), el nefroma quístico del adulto y el nefroma quístico pediátrico. Los nefromas quísticos del adulto son completamente quísticos, mientras que los MEST contienen un componente sólido variable. El nefroma quístico pediátrico es una neoplasia exclusivamente quística con mutaciones en DICER1, en la que los septos contienen únicamente tejido fibroso y túbulos diferenciados; a diferencia del nefroma quístico pediátrico, los nefromas quísticos del adulto normalmente contienen bandas de colágeno y son positivos para inhibina (WHO Classification of Tumours. Urinary and male genital tumours [Internet]. Lyon (France): IARC. 5th ed. 2022; vol. 8. Disponible en: https://tumourclassification.iarc.who.int/chapters/36 - disponible sólo con suscripción).
Nefroma quístico del adulto y tumor epitelial y estromal mixto (MEST)
El nefroma quístico (NQ) es una neoplasia quística benigna poco frecuente del riñón. Este tumor ha sido descrito con diversos nombres: nefroma quístico (el término usado por la WHO, en el grupo MEST, en la clasificación del 2022), nefroma multiquístico, tumor quístico multilocular, quiste multilocular renal, nefroma quístico multilocular, cistadenoma renal y riñón poliquístico parcial (Sharma A, et al. Asian J Urol. 2017;4:128-130. [PubMed link]).
Fue descrito por primera vez en 1892 como adenoma quístico del riñón. Su etiología e histogénesis es discutible, y en el pasado se consideraban lesiones del desarrollo con potencial maligno. El nombre de nefroma multiquístico se propuso por primera vez en 1951 y luego se modificó y se subdividió en nefroma quístico y nefroblastoma quístico parcialmente diferenciado de acuerdo a la ausencia o presencia de elementos blastemales, respectivamente. El NQ es una lesión quística benigna e infrecuente del riñón con distribución de edad bimodal, que se presenta tanto en niños como en adultos. Se observa con mayor frecuencia en menores de 4 años, con leve mayor frecuencia en hombres que en mujeres (nefroma quístico pediátrico - ver más adelante) y en adultos en la 4ª - 6ª décadas de la vida, con una relación hombre:mujer de 1:7. El nefroma quístico de inicio en adultos es histogenética y morfológicamente diferente del nefroma quístico pediátrico. El MEST ha sido llamado hamartoma de la pelvis renal y nefroma mesoblástico del adulto, pero, no tiene relación con éste ni presenta la misma alteración genética [t(12;15)(p13;q25)]. Se localiza habitualmente en médula o pelvis renal, bien circunscrito pero no encapsulado, color café o marrón. Se caracteriza por una mezcla de elementos epiteliales y estromales con patrones de crecimiento sólido y quístico. No hay blastema ni elementos epiteliales inmaduros; no hay atipia ni suelen verse mitosis (variante clásica). Puede haber una variante celular con más celularidad, mitosis y hemorragia. De acuerdo con la clasificación de neoplasias renales de la Organización Mundial de la Salud (OMS) (2022), el nefroma quístico del adulto se agrupa con el MEST. El término "familia de tumores epiteliales y estromales mixtos" se utiliza en la clasificación de la OMS para abarcar el nefroma quístico del adulto y el MEST; en esta clasificación, los casos pediátricos se denominan: "nefroma quístico pediátrico" y los casos en adultos: "nefroma quístico del adultos" (WHO Classification of Tumours. Urinary and male genital tumours [Internet]. Lyon (France): IARC. 5th ed. 2022; vol. 8. Disponible en: https://tumourclassification.iarc.who.int/chapters/36).
Macroscópicamente la lesión es bien definida, encapsulada, confinada al riñón, de tamaño variable y puede abultar hacia la superficie externa del órgano. El riñón adyacente es normal, salvo por la compresión. Los MEST y los nefromas quísticos son tumores bien delimitados y típicamente no encapsulados. Los nefromas quísticos son completamente quísticos, mientras que los MEST son sólidos y quísticos o completamente sólidos. Las zonas sólidas tienen una superficie de corte blanca y firme.
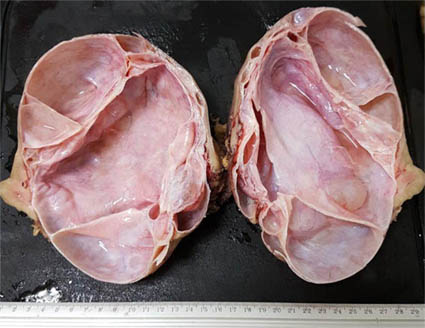
Figura 31a. Lesión multiquística en una mujer de 36 años. Obsreve las paredes delgadas, con aspecto fibroso, de esta lesion multiloculada. Histológicamente corresponde a un nefroma quistico multilocular (ver fotos microscópicas 31b y 31c).
Microscópicamente Los nefromas quísticos del adulto son enteramente quísticos, mientras que los MEST contienen un componente sólido variable. Tanto los nefromas quísticos como los MEST muestran quistes y glándulas de tamaño variable separados por estroma. El epitelio que recubre los quistes en el nefroma quístico suele ser plano, cúbico o en tachuela; raramente se han observado células con citoplasma claro. El estroma septal es de espesor variable, generalmente delgado, y está formado por tejido fibroso que varía de hipocelular a hipercelular, a veces parecido al estroma ovárico. Las calcificaciones estromales son comunes. Puede producirse luteinización estromal y nódulos de estroma hialinizado con contornos definidos que se asemejan a los cuerpos albicans ováricos. Rara vez se encuentran mitosis dispersas, pero no debe haber atipia ni necrosis. El componente epitelial en MEST es variable en arquitectura y tipo de célula. Las glándulas suelen estar dispersas en el estroma o muy agrupadas. Se han informado pequeños quistes que contienen material eosinofílico que recuerda a los folículos tiroideos, túbulos ramificados complejos, arquitectura de tipo filoides y estructuras papilares. También se ha descrito epitelio con aspecto urotelial, ciliado o con citoplasma claro o eosinófilo. Se han descrito células caliciformes mucinosas y diferenciación endometrioide. Son comunes en el estroma músculo liso y grasa. Pueden ocurrir cambios mixoides y edematosos. Rara vez puede ocurrir una transformación maligna, que generalmente se manifiesta como sarcomas que surgen del estroma. Inmunohistoquímica: el componente estromal suele ser positivo para actina, desmina, CD10, receptores estrogénicos y progestágenos y, con frecuencia, las células estromales luteinizadas expresan inhibina y calretinina. La catepsina K es negativa. El componente epitelial es PAX2/8 positivo. La inmunohistoquímica es útil para excluir el sarcoma sinovial renal, el tumor papilar de células claras y el carcinoma de células renales tubuloquísticos (el anterior texto fue tomado de: WHO Classification of Tumours. Urinary and male genital tumours [Internet]. Lyon (France): IARC. 5th ed. 2022; vol. 8. Disponible en: https://tumourclassification.iarc.who.int/chapters/36). [Imagen micro (link)] [Otras más (link)]

Figura 31b. Las paredes están formadas por un estroma fusocelular benigno. La microscopía electrónica. Las cavidades pueden estar tapizadas por epitelio simple que puede verse plano, cúbico o en "tachuela" (H&E, X100).
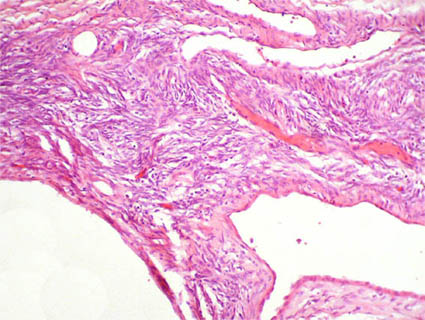
Figura 31c. A mayor aumento el estroma recuerda el aspecto de estroma ovárico. Puede verse un epitelio cúbico simple recubriendo algunas de las cavidades (H&E, X200).
Es una lesión completamente benigna, no metastatizante. La resección quirúrgica es el método eficaz para excluir las lesiones quísticas malignas del riñón. Sin embargo, la cirugía conservadora de nefronas (nefrectomía parcial) puede ser una opción según el sitio y el tamaño de la lesión (Wilkinson C, et al. Adult multilocular cystic nephroma: Report of six cases with clinical, radio-pathologic correlation and review of literature. Urology Annals. 2013;5:13-17. [PubMed link]).
Ver el Caso 143 de nuestra serie de casos: Nefroma Quístico Multilocular.
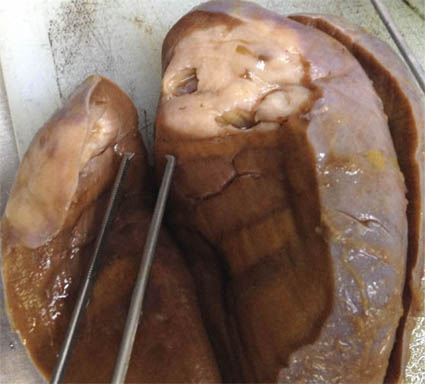
Figura 31d. Tumor renal sólido con cavidades en una mujer de 44años. La histología demostró ser un tumor estromal y epitelial mixto. Ver las imágenes micro más adelante.
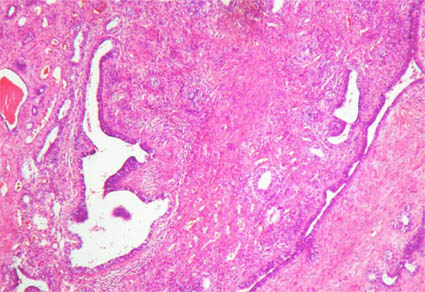
Figura 31e. Mescla de un estroma fusocelular benigno con elementos epiteliales (H&E, X100).
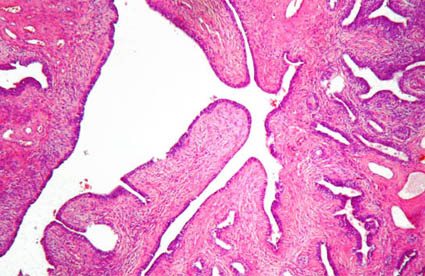
Figura 31f. Áreas estromales menos celulares y con proyecciones hacia una pequeña cavidad, con componente epitelial de altura variable (H&E, X100).

Figura 31g. A mayor aumento vemos túbulos con aspecto primitivo en medio de un estroma moderadamente celular, benigno, sin atipias (H&E, X200).
Diagnóstico diferencial: Con carcinoma sarcomatoide: estos tienen más atipia, mitosis y necrosis. Con nefroblastoma y nefroblastoma parcialmente diferenciado: éstos tienen blastema. También podemos plantearnos problemas diagnósticos con leiomioma, tumor fibroso solitario, angiomiolipoma y adenofibroma metanéfrico. (Picken MM, Fresco R. Mixed epithelial and stromal tumor of the kidney: preliminary immunohistochemical and electron microscopic studies of the epithelial component. Ultrastruct Pathol 29:283-6, 2005. [PubMed link]; Hora M, et al., Extensively Cystic Renal Neoplasms in Adults (Bosniak Classification II or III) - Possible "Common" Histological Diagnoses: Multilocular Cystic Renal Cell Carcinoma, Cystic Nephroma, and Mixed Epithelial and Stromal Tumor of the Kidney. Int Urol Nephrol 37:743-50, 2005 [PubMed link]).
Ver el Caso 98 de nuestra serie de casos (tumor estromal y epitelial mixto en una mujer de 44 años).
El nefroma quístico pediátrico es una neoplasia exclusivamente quística con mutaciones en DICER1, en la que los septos contienen únicamente tejido fibroso y túbulos diferenciados. La presentación típica es la de una masa abdominal, ocurre con mayor frecuencia en niños que en niñas y la mayoría de los pacientes tienen <24 meses. La mayoría de los nefromas quísticos pediátricos están bien delimitados microscópicamente por una pseudocápsula fibrosa, pero una minoría carece de pseudocápsula, ya sea parcial o completamente. Están compuestos enteramente por quistes septados revestidos por epitelio aplanado, cúbico o en tachuela, o están denudados. Los tabiques contienen tejido fibroso con condensaciones celulares focales y túbulos bien diferenciados. El estroma marca para receptores estrogénicos como muchas otras lesiones renales quísticas. La presencia de elementos nefroblásticos inmaduros excluye el diagnóstico de nefroma quístico pediátrico e indica nefroblastoma quístico parcialmente diferenciado. Las lesiones conocidas como nefromas quísticos del adulto pertenecen a la familia de tumores mixtos epiteliales y estromales; a diferencia del nefroma quístico pediátrico, normalmente contienen bandas de colágeno y son positivos para inhibina. La demostración de la mutación DICER1 es útil en algunos casos. El nefroma quístico pediátrico se trata únicamente con cirugía. Su pronóstico es excelente, con supervivencia global libre de eventos del 100%. Se han descrito casos muy raros de transformación del nefroma quístico en sarcoma DICER1, pero el riesgo se considera muy bajo (el anterior texto fue tomado de: WHO Classification of Tumours. Urinary and male genital tumours [Internet]. Lyon (France): IARC. 5th ed. 2022; vol. 8. Disponible en: https://tumourclassification.iarc.who.int/chapters/36).
Ver el Caso 214 de nuestra serie de casos (nefroma quístico pediátrico en un niño de 2 años).
Sarcoma sinovial: Es un tumor que muy raras veces se origina en el riñón. Las caractetrísticas morfológicas y el pronóstico es similar al de otras localizaciones. Hay una mezcla de componentes epiteliales y estromales, aunque hay casos monofásicos difíciles de diagnosticar sin inmunohistoquímica (CK positivos).
Otros tumores que podemos encontrar en riñones de adultos son los mencionados en la Tabla 1 e incluyen: leiomiosarcoma (algunos de la vena renal), angiosarcoma, rabdomiosarcoma, histiocitoma fibroso maligno, hemangiopericitoma, osteosarcoma, leiomioma, hemangioma, linfangioma, schwanoma, tumor fibroso solitario, carcinoide, carcinoma neuroendocrino, tumor neuroectodérmico primitivo, neuroblastoma, feocromocitoma, neoplasias hematolinfoides, teratomas, coriocarcinomas y lesiones de origen metastásico.
Ver el Caso 162 de nuestra Serie de Casos: Angiosarcoma Renal Primario.
Tumores renales pediátricos
Nefroblastoma (tumor de Wilms) (Se ha recomendado llamarlo sin el posesivo: tumor Wilms)
Los nefroblastomas son derivados de blastema renal embrionario y recapitulan varios estadios del desarrollo del órgano. Es el tumor renal maligno más frecuente en niños y ocupa el 5º lugar en frecuencia entre los tumores malignos de la infancia (después de leucemias, linfomas, tumores cerebrales y neuroblastomas). En muchos casos se identifican nidos de tejido inmaduro: nefroblastomatosis, acompañando a esta neoplasia. Afecta 1 por 8-10.000 niños.
Es un tumor predominantemente de la infancia, 90% de los casos se producen antes de los 6 años de edad; es inusual en los primeros 6 meses de vida y ocurre esporádicamente en adultos. En la mayoría de casos se presentan como una masa palpable; también pueden presentarse con hematuria, dolor abdominal, obstrucción gastrointestinal, hipertensión secundaria y con síntomas por ruptura traumática de la masa. No hay predilección por sexos y tiene una distrubución mundial más o menos homogénea. Son más frecuentes en afroamericanos y menos frecuentes en asiáticos. En la mayoría de casos es esporádico, pero puede presentarse en el contexto de enfermedades genéticas como el síndrome WAGR (acrónimo en inglés de Wilms, aniridia, anormalidades genitales y retardo mental); síndrome de Beckwith-Wiedemann (onfalocele, macroglosia, hemihipertrofia, quistes renales medulares, citomegalia renal); hemihipertrofia; síndrome de Denys-Drash (nefroblastoma, pseudohermafroditismo, glomerulonefritis). Además hay casos de nefroblastoma familiar.
El pronóstico del nefroblastoma es bueno en estadios iniciales, sin anaplasia y edad menor de 4 años; en la actualidad hay esquemas de quimioterapia que han demostrado muy buenos resultados y altos porcentajes de curación. Los sitios más comunes de metástasis son ganglios linfáticos, pulmones e hígado; pocas veces dan metástasis al sistema nerviosos central o a huesos. Hay una supervivencia a largo plazo cercana al 90%.
Entre los factores pronósticos favorables están: edad menor de 2 años, estadio I, especimen de nefrectomía <550 g., ausencia de anaplasia.
Hallazgos macroscópicos: Son lesiones únicas y unilaterales en la mayoría de casos, usualmente tienen gran tamaño: >5 cm o >500g.; son masas sólidas, bien circunscritas, a veces con una pseudocápsula; tienen consistencia blanda, friable, son de color crema o marrón, con áreas de hemorragia y necrosis que puede producir cavidades quísticas. En algunos casos hay septos intratumorales que le dan un aspecto lobulado. La lesión suele comprimir el resto del riñón y podemos ver extensión a la grasa perirrenal y/o del hilio. En algunos casos la lesión protruye a cavidades pielocaliciales. El tumor es bilateral en <5% de casos y puede ser multicéntrico.
Hallazgos microscópicos: Es una neoplasia con gran variabilidad histológica, pero, hay tres componentes básicos que se encuentran en variable proporción: blastemal, epitelial y mesenquimal.
Componente blastemal:
Compuesto por células pequeñas, indiferenciadas, con núcleos
hipercromáticos, cromatina burda, nucléolos poco notorios o no
visibles y escaso citoplasma; se disponen en láminas sólidas,
nidos, cordones o adoptando un patrón basaloide. Hay actividad mitótica
prominente.
Componente epitelial: Estructuras tubulares y glomerulares
con aspecto primitivo (embrionario), a veces con formaciones papilares y rosetas,
podemos ver, en algunos casos, diferenciación escamosa, neural o endocrina.
Componente mesenquimal: Tiene aspecto fibroblástico,
mixoide o fibromixoide; menos comunmente se observa diferenciación a
músculo esquelético, músculo liso, hueso. cartílago,
tejido adiposo o tejido neuroglial (elementos heterólogos).
La mayoría de nefroblastomas muestran los tres componentes; si hay uno de ellos que forma más del 66% del tumor lo designamos de acuerdo a este (blastemal, epitelial o mesenquimal). Algunos casos son bifásicos y ocasionalmente monofásicos. En casos muy inusuales encontramos diferenciación difusa a músculo esquelético (nefroblastoma rabdomiomatoso fetal) o encontramos combinaciones de elementos epiteliales y estromales maduros (nefroblastoma teratoide). [Varias imágenes macro y micro (link)]

Figura 32. Tumor de Wilms en el que se identifican dos componente: epitelial, formando túbulos o con aspecto en "seudorrosetas" (o "pseudotúbulos"), y blastemal: células con poco citoplasma, densamente agrupadas, sin diferenciación. (H&E, X200)
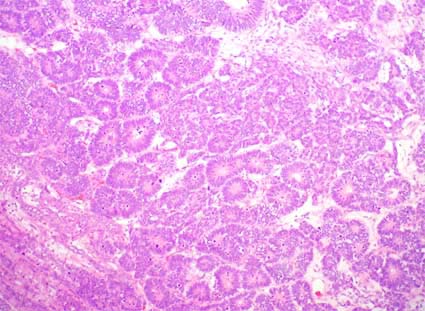
Figura 33. Componente epitelial de tumor de Wlms; es el mismo caso de la figura 30. Aquí vemos el aspecto "rosetoide" más detallado. El componente epitelial es muy variable, puede haber túbulos primitivos o bien diferenciados, estructuras glomeruloides y otros aspectos que recuerdan elementos epiteliales primitivos. (H&E, X100)
En la evaluación histológica
es muy importante la valoración de anaplasia nuclear para determinar
histología "favorable" o "no favorable".
Histología no favorable: Se define por la presencia
de núcleos hipercromáticos que son al menos tres veces más
grandes que los núcleos del mismo tipo celular; este aumento debe verse
en los tres ejes, es decir, no debe ser simplemente elongación. Células
multinucleadas y núcleos grandes en rabdomioblastos no indican anaplasia
a menos que tengan mitosis atípicas. Figuras mitóticas multipolares
que aperecen hiperploides y grandes; este tamaño grande indica aumento
del ADN. Deben estar presentes tanto el hipercromatismo y aumento marcado del
tamaño nuclear como las figuras mitóticas multipolares para cumplir
los criterios de anaplasia. La anaplasia puede ser focal o difusa:
Anaplasia focal: Debe cumplir todos los siguientes
requisitos: a.) la anaplasia esta limitada al tumor primario y no está
presente en áreas de invasión o compromiso extrarrenal; b.) no
hay atipia citológica "significativa" (que no llena los requisitos
de anaplasia) en otras áreas diferentes a los focos de anaplasia; y c.)
los focos de anaplasia están claramente demarcados de las zonas adyacentes
no anaplásicas. El tamaño de los focos de anaplasia no es importante
siempre que estén localizados; pueden haber múltiples focos de
anaplasia, pero deben cumplir los tres requisitos mencionados.
Anaplasia difusa: Está presente una o más
de las siguientes características: a.) anaplasia no localizada; b.) tumor
anaplásico en vasos intra o extrarrenales; c.) anaplasia fuera del riñón;
d.) anaplasia en tejido de una biopsia pequeña tomada de un sitio aleatoriamente
determinado; e.)anaplasia que se extiende hasta el borde de uno o más
cortes (los bordes de la zona o foco anaplásico no pueden determinarse
con claridad); y f.) la anaplasia está asociada con atipia nuclear severa
(que no llena los requisitos de anaplasia) en otros sitios del mismo tumor (Beckwith
JB, National Wilms Tumor Study: An update for pathologists. Pediatr Dev Pathol
1:79-84, 1998
[PbMed
link]). [Varias imágenes micro, incluyendo anaplasia (link)]

Figura 34. Tumor de Wilms, componente epitelial en medio del cual se identifican células grandes, muy llamativas, con mitosis claramente atípicas, multipolares, indicativas de anaplasia. Es necesario evaluar la extensión y localización de las áreas con anaplasia para determinar si es focal o difusa (ver texto). (H&E, X300).
La anaplasia está asociada con mayor resistencia a la quimioterapia y no es un marcador de agresividad tumoral. Anteriormente se llamaba anaplasia focal a los tumores con <10% de campos microscópicos conteniendo características anaplásicas.
La IHQ generalmente no aporta datos adicionales en el estudio de nefroblastomas. El componente blastemal es positivo para vimentina pero no suele mostrar marcadores que indiquen alguna diferenciación. El componente mesenquimal puede ser positivo para algunos marcadores de acuerdo al componente, p.e. marcadores musculares si se diferencia a músculo.
Por microscopía electrónica el tumor recuerda el metanefros en desarrollo, con uniones celulares bien desarrolladas, microvellosidades y una capa de material grueso que rodea las células.
Diagnóstico diferencial: Incluye una gran cantidad de lesiones neoplásicas de células pequeñas que semejan mucho el componente blastemal o aun epitelial: nefroblastomas, tumor neuroectodérmico primitivo (PNET), tumor de células pequeñas redondas desmoplásico intraabdominal, linfoma, sarcoma de células claras y, algunas veces, carcinoma de células renales. En el diagnóstico diferencial es muy importante la localización y la presentación clínica. Las metástasis pulmonares sugieren un nefroblastoma más que un neuroblastoma; los nefroblastomas pocas veces metastatizan a hueso, contrario al sarcoma de células claras. La IHQ es muy útil en el diagnóstico de neuroblastomas, tumor desmoplásico intraabdominal de células pequeñas y redondas, y linfomas.
Estudios moleculares demuestran expresión anormal del gen WT1 (11p13, que codifica un factor de transcripción expresado en el sistema urogeniltal embrionario) y WT2 (11p15.5); también se han descrito alteraciones en cromosomas 1, 7q, 8, 12 y 16; hay anormalidades del gen p53 en focos de anaplasia. No hay la translocación (12;15) descrita en el nefroma mesoblástico celular ni t(11;22) del Ewing/PNET, ni alteraciones del 22q11.2 del tumor rabdoide ni del N-myc como en el neuroblastoma.
Factores indicadores de mal pronóstico:
1.) Anaplasia nuclear; la anaplasia difusa indica peor pronóstico que
la focal, pero aun pequeños focos se relacionan con resistencia a la
quimioterapia.
2.) Estadio alto. La mayoría de tumores predominantemente epiteliales
están en estadio I y la mayoría de predominantemente blastemales
se encuentran en estadio III/IV.
3.) Edad mayor de 2 años.
4.) Expresión de P-glicoproteína en células endoteliales
(Camassei FD et al., Am J Clin Pathol 17:484-90,
2002 [PubMed
link])
5.) Presencia de pseudocapsula inflamatoria, invasión del seno renal
o de vasos intrarrenales
6.) Gran tamaño de la masa tumoral.
Estadificación del tumores renales pediátricos (National Wilms Tumor Study Group)
Estadio I: Tumor limitado al riñón y completamente resecado. Se requiere que a.) la cápsula renal esté intacta (no penetrada por el tumor); b.) no haya invasión de vasos venosos o linfáticos; y c.) que no haya tumor residual o compromiso de ganglios linfáticos. Puede haber invasión pequeña de los tejidos blandos del seno renal pero los márgenes de resección mediales deben estar libres de tumor.
Estadio II: El tumor se extiende más allá del riñón, pero se ha resecado completamente, con márgenes negativos y sin compromiso de ganglios linfáticos. Se diagnostica este estadio en cualquiera de las siguientes situaciones: a.) el tumor penetra la cápsula renal o compromete vasos del seno renal, pero, con márgenes quirúrgicos negativos para compromiso tumoral; b.) invasión vascular fuera del parénquima renal; c.) caída de tejido tumoral en el flanco durante la cirugía (pero no contaminación peritoneal; d.) el tumor invade más que mínimamente los tejidos blandos del seno renal.
Estadio III: Tumor residual confinado al abdomen, sin diseminación hematógean. Incluye casos con cualquiera de las siguientes características: a.) tumor residual en abdomen; b.) tumor en ganglios linfáticos abdominales; c.) contaminación peritoneal difusa por crecimiento tumoral directo, implantes o derrame durante el acto quirúrgico; d.) márgenes quirúrgicos positivos, e.) historia de biopsia del tumor antes de la cirugía definitiva.
Estadio IV: Metástasis hematógenas (incluyendo órganos intraabdominales) o a ganglios linfáticos extraabdominales (o más allá de la región de drenaje del riñón).
Estadio V: Nefroblastoma bilateral. (cada tumor debe estadificarse separadamente de I a IV).
Nefroblastoma quístico parcialmente diferenciado
Son lesiones que, según algunos autores, podrían evolucionar de nefroblastoma o de restos nefrogénicos, aunque hay controversia respecto a su histogénesis. Se le suele relacionar también con el nefroma quístico. Hay dos picos de edad en la que se presentan: niños menores de dos años y adultos de edad media o mayores; en los primeros hay predominancia masculina y en los segundos femenina. Muchos casos son asintomáticos, pero, pueden presentarse como masa o hematuria. Son lesiones benignas pero pueden recurrir si no son completamente resecados.
Son usualmente unilaterales y solitarios, miden 5-10 cm en promedio y macroscópicamente son bien circunscritos, multiquísticos y sin hemorragia o necrosis. Las cavidades son de tamaño variable y están separadas por septos fibrosos, sin masas o nódulos expansivos (si es así debe plantearse la presencia de un nefroblastoma o de un carcinoma [en adultos]).
Hallazgos microscópicos: Las cavidades quísticas están revestidas por epitelio cúbico simple que puede estar aplanado por compresión, su citoplasma es esosinofílico y sus núcleos no muestran atipia. Los septos están formados por tejido fibroso y contienen nidos de blastema, sin formar masas sólidas expansivas (sería entonces un nefroblastoma). La diferenciación con el nefroma quístico está en que éste no contiene nidos de blastema.
En algunos casos podemos ver células claras en las cavidades, pero no debe haber nódulos en los septos. Ocasionalemnte hay elementos heterólogos (músculo y grasa principalmente).
Restos nefrogénicos (y nefroblastomatosis)
Son focos anormales de tejido embrionario nefrogénico; cuando son múltiples se les llama nefroblastomatosis. Se encuentran en aproximadamente el 1% de autopsias de niños pequeños; cuando hay nefroblastoma se detectan en hasta el 40% de los casos. La presencia de restos nefrogénicos en riñones con nefroblastoma indica un riesgo incrementado de esta neoplasia en el riñón contralateral.
Se ha propuesto una clasificación de acuerdo con su localización y morfología (Beckwith et al., Pediatr Pathol 10:1-36, 1990 [PubMed link]):
Restos nefrogénicos perilobares:
En la periferia de los lóbulos, compuestos predominantemente de túbulos
y blastema, con escaso estroma; a menudo únicos y claramente demarcados.
Intralobares: Localizados en la corteza y médula;
con frecuencia predomina el componente estromal sobre el blastemal y tubular;
se mezcla irregularmente con el tejido renal y es, a menudo, multifocal.
Nefroblastomatosis combinada: Hay restos nefrogénicos
perilobares e intralobares.
Nefroblastomatosis universal: Una variante rara en
la que el tejido renal consiste, enteramente, de estructuras embrionarias.
Además, los restos nefrogénicos son subclasificados de acuerdo con su desarrollo y apariencia morfológica como: latentes, incipientes, en involución (esclerosantes), obsolescentes, hiperplásicos y neoplásicos (Beckwith et al., Pediatr Pathol 10:1-36, 1990 [PubMed link]).
También llamado "tumor renal de la infancia metastatizante a hueso". Es una neoplasia poco frecuente que tiene mucha tendencia a metastatizar a huesos. Su histogénesis es desconocida; podrían diferenciarse hacia mesénquima nefrogénico primitivo. Corresponde al 4-5% de neoplasia renales pediátricas. Se suelen presentar entre los 1 y 3 años de edad, son muy raros antes de los 6 meses y no se describen casos en adultos. Hay predominancia en el sexo masculino (3:2). No hay predominancia de alguna raza, no se asocian con enfermedades genéticas y no parecen haber casos familiares. Es muy metastatizante y, además de huesos, pueden haber metástasis a ganglios linfáticos, cerebro, pulmón, hígado y tejidos blandos.
El tratamiento incluye cirugía y quimioterapia; su tratamiento es diferente al del nefroblastoma ya que son resistentes a esta quimioterapia. En muchos casos hay recurrencias tardías, después de varios años.
Hallazgos microscópicos: Son tumores formados por una proliferación sólida de células con núcleos redondeados, con cromatina fina, pequeños nucléolos y citoplasma pálido con límites mal definidos. El citoplasma tiene un aspecto vacuolado claro o finamente granular debido a la presencia de mucopolisacáridos. No es universal el aspecto claro del citoplasma, algunas células lo tienen debilmente eosinofílico. A pesar de su nombre la carcaterística de citoplasma claro es prominente en apenas un 20% de casos. Las células forman cordones y nidos separados por delgados septos que contienen pequeños vasos sanguíneos ramificados. La actividad mitótica es variable, en algunos casos muy baja comparativamente con otros tumores pediátricos. La neoplasia es mal delimitada y presenta bordes infiltrativos que se mezclan con el parénquima adyacente englobando túbulos y glomérulos. El englobamiento de túbulos aislados es muy común y útil para el diagnóstico en esta neoplasia. Algunas veces estos túbulos atrapados presentan cambios celulares reactivos con aspecto displásico, este hallazgo puede hacer confundir con el componente epitelial de un tumor de Wilms (Figura 36). [Múltiples imágenes microscópicas en la presentación de un bonito caso (link)]
![]()
Figura 35. Sarcoma renal de células claras. Observe el patrón difuso, mal diferenciado de la neoplasia. En muchas áreas las células no son realmente claras y presentan un citoplasma escaso y/o eosinófilo. (H&E, izquierda, X300; derecha, X400)
![]()
Figura 36. La presencia de túbulos atrapados, aislados en medio de la neoplasia es un hallazgo útil en el sarcoma renal de células claras. Cuando se trata del componente epitelial de un tumor de Wilms, estas estructuras no suelen verse aisladas y habitualmente hay conjuntos de estructuras tubulares o epiteliales de otro tipo. Observe el aspecto epitelial que recuerda un epitelio "displásico". (H&E, X200)
Hay algunas variantes histológicas: de células fusiformes; otros con prominentes bandas de colágeno que le dan un aspecto esclerosante; mixoide; quistico; epitelioide; formando empalizada; patrón sinusoidal similar a hemangiopericitomas; y tumores con áreas pleomórficas. Estos patrones pueden combinarse con áreas de aspecto "clásico".
No hay marcadores de IHQ específicos y esta técnica es más útil para excluir otros tumores. Por ultraestructura hay escasos filamentos intermedios y uniones intercelulares incompletas. Recientemente se ha descrito, en sólo algunos casos, una translocación que origina un gen de fusión: t(10:17) (q22;p13), pero no se conoce el papel de este gen (O'Meara E, et al. Characterization of the chromosomal translocation t(10;17)(q22;p13) in clear cell sarcoma of kidney. J Pathol. 2012;227(1):72-80. [PubMed link])
Diagnóstico diferencial: Con nefroblastoma: no se evidencia blastema ni componentes mesenquimales heterólogos en el sarcoma de células claras. Con el tumor rabdoide: no hay, en el sarcoma de células claras, los núcleos prominentes y el citoplasma abundante del tumor rabdoide; en ocasiones pueden presentar áreas parecidas a las del tumor rabdoide, pero, en general, habrá áreas más características del sarcoma de células claras. Las variantes morfológicas pueden llevar a plantearse otras opciones en el diagnóstico diferencial, en estos casos un muestreo generoso del tumor revelará áreas más típicas con células de citoplasma pálido, apariencia homogénea y tendencia a englobar estructuras del parénquima renal.
Ver el Caso 75 de nuestra serie de casos: Sarcoma de células claras
Es un tumor poco frecuente con histogénesis desconocida; corresponde a menos del 2% de tumores renales de la infancia. Alrededor del 90% de casos ocurren antes de los 2 años de edad y son muy inusuales después de los 4 años. Son algo más frecuentes en hombres (1,5:1). Suelen haber metástasis al momento del diagnóstico y se presentan como masas grandes intraabdominales. En aproximadamente el 15% de casos se asocian con tumores embrionarios de la linea media, principalmente meduloblastoma.
El tumor rabdoide es el más maligno de todas las neoplasias renales pediátricas, son extensamente metastatizantes y suelen causar la muerte en los primeros 12 meses después del diagnóstico.
Se describe también un tipo de tumor, llamado de igual manera, en sitios extrarrenales (tejidos blandos) pero con un rango de edad de presentación mucho más amplio. También es relativamente frecuente encontrar áreas con aspecto rabdoide en carcinomas renales (hasta en el 5%), se asocia e tumores grado 3 y 4 de Fuhrman, estadio avanzado y se relacionan con peor pronóstico y alta mortalidad; en estos casos no se trata de un tipo de tumor, sino, de un aspecto morfológico en diferentes carcinomas; debemos plantearnos esta posibilidad en casos de tumores con aspecto rabdoide (así sea difuso) y edad mayor de 5 años.
Hallazgoa macroscópicos: Estos tumores son blandos, carnosos, grisáceos o con tono amarillo, usualmente grandes, no encapsulados, con lesiones satélite, hemorragia y necrosis. Generalmente se ubican en la porción media del órgano y comprimen o invaden la pelvis renal.
Hallazgos microscópicos: Hay un crecimiento sólido de una población de células grandes, monótonas, poco cohesivas, con núcleos grandes y excéntricos, con nucléolos prominentes y citoplasma eosinofílico abundante que contiene una típica inclusión hialina grande que desplaza el núcleo; estas inclusiones no las vemos en todas las células, pero, son prominentes y fáciles de encontrar. Con el patrón clásico pueden combinarse otros patrones: esclerosante, epitelioide, fusiforme, linfomatoide, vascular, pseudopapilar y quístico.
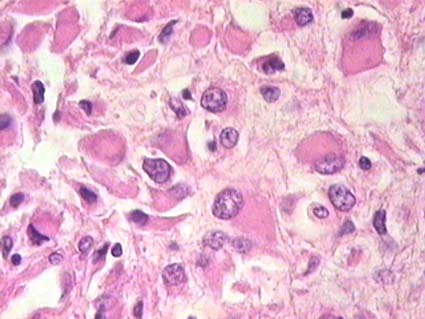
Figura 37. Tumor rabdoide. Observe las prominentes inclusiones intracitoplasmáticas, eosinofílicas, que carcaterizan a estos tumores. Células similares pueden verse, ocasionalmente, en carcinomas renales. (H&E, X400).
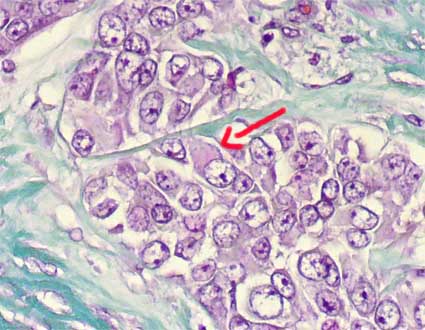
Figura 38. Esta imágen fue obtenida de un campo en un carcinoma convencional de un paciente de 59 años. Hay células rabdoides (flecha), con núcleos de alto grado. En algunos casos este cambio rabdoide es extenso, haciendo plantear la posibilidad de un tumor rabdoide. En mayores de 5 años de edad, no debemos pensar en tumor rabdoide, sino, en carcinomas con áreas rabdoides. (Tricrómico de Gomori, X400).
Por IHQ hay resultados contradictorios en la literatura, pero son usualmente positivos para vimentina, citoqueratinas y EMA demostrándose su origen epitelial. Son usualmente negativos con marcadores musculares, neurales y CD99. En casos de carcinomas renales con características rabdoides encontraremos expresión de los siguientes marcadores (porcentajes de positividad aproximado): Vim (100%), EMA (47%), panCK (56%) NSE (79%), S-100 (37%); y negatividad para CK7, CK20, actina de músculo liso y actina sarcomérica, desmina, HMB45 y proteína ácida fibrilar glial.
Por microscopía electrónica se evidencian inclusiones citoplasmáticas arremolinadas que corresponden a agregados de filamentos intermedios de 6-10 nm de grosor.
No hay anormalidades citogenéticas consistentemente expresadas por estos tumores.
Diagnóstico diferencial: El principal problema diagnóstico se plantea con otros tumores que pueden presentar carcaterísticas rabdoides y que incluyen: nefroblastoma, carcinoma de células renales (cualquiera de los tipos), nefroma mesoblástico, carcinoma urotelial, carcinoma medular renal y rabdomiosarcoma; un amplio muestreo de estos tumores mostrará el aspecto carcaterístico de cada uno; es muy importante también correlacionar con la edad y tener presente que el tumor rabdoide se presenta en menores de 5 años y lo diagnosticamos luego de estudiar múltiples cortes del tumor.
Nefroma mesoblástico congénito
También se le ha llamado hamartoma leiomiomatoso, un nombre que parece incorrecto dada su naturaleza neoplásica. Es una neoplasia renal congénita usualmente detectada antes de los 6 meses de edad, aunque se han descrito casos en adultos. Se suelen presentar como masas intraabdominales y ocasionalmente se relacionan con polihidramnios y parto prematuro.
Son lesiones que suelen curarse con la resección quirúrgica y no necesitan radio o quimioterpia. En pocos casos se han descrito metástasis a distancia y en hasta el 7% hay recurrencia local que puede producir invasión al retroperitoneo y complicaciones que pueden ser fatales. No hay criterios histológicos que se correlacionen consistentemente con el pronóstico y, al parecer, la edad (menor de 3 meses) y una adecuada resección completa, son los mejores indicadores pronóstico.
Macroscópicamente son masas sólidas, grisáceas, amarillas o marrones, con aspecto arremolinado que recuerda los leiomiomas uterinos. Los límites pueden ser nitidos o pueden presentar un aspecto infiltrativo. En algunos casos hay necrosis y hemorragia. Muchos casos están ubicados cerca del hilio y pueden comprimirlo; en algunos casos hay extensión más allá de la cápsula renal.

Figura 38a. Imágenes macroscópica característica de nefroma mesoblástico congénito. Este caso corresponde al de un niño de 4 meses de edad, con diagnóstico de masa renal desde el segundo mes de vida.
Hallazgos microscópicos: La característica principal es proliferación de células fusiformes con un grado de celularidad variable. Esta células tienen características de mesénquima secundario, que, en contraste con el mesénquima primitivo, no tienen la capacidad de formar elementos epiteliales. Las células en proliferación tienen características de fibroblastos, miofibroblastos o de músculo liso. La neoplasia puede rodear túbulos y glomérulos y en algunos casos podemos encontrar cartílago y hematopoyesis extramedular. La cantidad de mitosis es variable pero suele aumentar en paralelo con el grado de celularidad. Hay un patrón clásico con baja celularidad y conteo mitótico que recuerda las fibromatosis (30%). Otro patrón es el descrito como variante celular (50%), en él la celularidad y número de mitosis es mayor; aquí se describen dos subtipos: i) de células gruesas y cortas ("rechonchas"): células más grandes, núcleo vesicular y nucléolo prominente, se suelen acompañar por áreas con células más fusiformes; y ii) el subtipo de "células azules": células más pequeñas, densamente agrupadas y con poco citoplasma, los nucléolos son inconspicuos. La variante cellular tiene algún riesgo de metástasis o recidiva (5-10%). Pueden combinarse los patrones clásico y celular (20%). [Varias imágenes (link)]
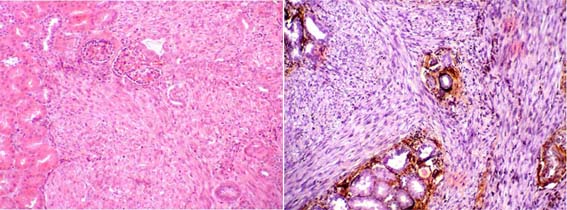
Figura 38b. Nefroma mesoblástico congénito, variante clásica. Proliferación de células fusiformes, con notoria celularidad aunque poco pleomorfismo; el conteo mitótico en casos de la variante clásica suele ser bajo. La lesión es infiltrativa, sin evidente cápsula, recordando la fibromatosis infantil. Izquierda: H&E, X200, derecha: plata-metenamina, X200.
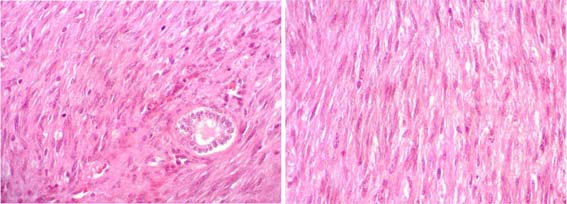
Figura 38c. A mayor aumento vemos el aspecto fibromatoso de la lesión, con poco pleomorfismo y englobando túbulos (izquierda). Igual caso que la figura anterior. Ambas imágenes: H&E, X400.
Por IHQ las células son positivas para vimentina, actina y desmina; la CK suele ser negativa en las células neoplásicas y resalta los componentes epiteliales atrapados en el tumor.
Citogenética: No hay las alteraciones en el cromosoma 11 descritos en el Wilms; se describen polisomías en los cromosomas 8, 11, 17 y 20. Recientemente se ha encontrado en estos tumores la translocación t(12;15)(p13;q25) que resulta en la fusión: ETV6-NTRK3, la misma alteración que se detecta en fibrosarcomas infantiles, sugieriendo que pueden tratarse de una misma neoplasia.
Diagnostico diferencial: Nefroblastoma: no hay blastema y no presenta diferenciación heteróloga. Sarcoma de céluals claras: puede ser un diagnóstico diferencial difícil; en el nefroma mesoblástico la celularidad puede ser más alta con superposición de núcleos, células más fusiformes y citoplasma menos claro; la positividad para desmina y actina es propia del nefroma mesoblástico.
Ver el Caso 48 de nuestra Serie de Casos: Nefroma mesoblástico congénito, variante clásica
Ver el Caso 138 de nuestra Serie de Casos: Nefroma mesoblástico congénico, variante celular
Tumor renal osificante de la infancia
Es un tumor muy raro con sólo algunos ejemplos informados en la literatura mundial. Se presentan como masas calcificadas en el sistema pielocalicial con bordes mal definidos en la médula renal. Microscópicamente está formado por áreas poco celulares con células de núcleos pequeños redondos u ovales y citoplasma pálido que se encuentran inmersas en una matriz de hueso denso. Hay muy pocas o ninguna mitosis detectable. No hay osteoclastos ni tejido cartilaginoso. Es una lesión benigna sin informes de recurrencia o metástasis. El tratamiento es la resección quirúrgica (Sotelo-Avila C, et al., Ossifying renal tumor of infancy: a clinicopathologic study of nine cases. Pediatr Pathol Lab Med 15:745-62, 1995 [PubMed link]; Steffens J, et al., Ossifying renal tumor of infancy. J Urol 149:1080-1, 1993 [PubMed link]).
Principales fuentes bibliográficas:
Bibliografía