Homepage - - - Tutorial Index
Amyloidosis, Multiple myeloma, Light chains deposition disease, Waldenström's macroglobulinemia, Monoclonal gammopathy of undetermined significance, Fibrillary and immunotactoid glomerulopathy and cryoglobulinemias
RENAL INVOLVEMENT IN MONOCLONAL GAMMOPATHIES
Monoclonal gammopathy signifies the presence of a monoclonal immunoglobulin (MIg) or its component. This is produced by a B-cell or plasma cell clone. When there is no end-organ damage, the condition is referred to as monoclonal gammopathy of undetermined significance (MGUS). MGUS is a premalignant condition that precedes all cases of multiple myeloma and immunoglobulin light chain (AL) amyloidosis. According to the physicochemical properties of the paraprotein, they can deposit, organize in different ways from the ultrastructural point of view, leak into urine and cause different types of tubular or interstitial damage or they can activate complement or pro-thrombotic factors and lead to great variability of clinical and morphological presentations.
Monoclonal gammopathy of renal significance (MGRS) is a heterogeneous group of kidney disorders that encompasses kidney lesions directly or indirectly caused by a nephrotoxic MIg. The concept of MGRS was introduced in 2012 by the International Kidney and Monoclonal Gammopathy Research Group (IKMG) to refer specifically to disorders with a MIg in the serum and/or urine in combination with kidney lesions, but in the absence of diagnostic criteria for hematological malignancy.
The toxicity of MIg is not limited to the kidney. As many organs can be affected, the term monoclonal gammopathy of clinical signifi- cance (MGCS) was introduced. Although all MGRS are also MGCS, some MGCS only have renal symptoms (pure MGRS), while others have both renal and other organ(s) damage.
Conceptually, monoclonal deposits in MGRS can affect any or all of the renal compartments. Therefore, one way of classifying MGRS is based on the dominant site of monoclonal deposition, althouht it is an arbitrary schema because most MGRS lesions involve more than one compartment within a single entity, and significant overlap exists; another method of classification relies on the histological and ultrastructural findings. Both classifications are pertinent: although the former is clinically relevant because it helps to clarify the pathogenesis and clinical features of MGRS, the latter is relevant from a diagnostic standpoint. Nevertheless, both schemes are commonly used together in clinical practice for better clinicopathological correlation (Jain A, et al. Pathophysiology and management of monoclonal gammopathy of renal significance. Blood Adv. 2019;3(15):2409-2423. [PubMed link]).
The list of entities in the spectrum of renal involvement by MIgs includes: by direct mechanism: cast nephropathy, amyloidosis (AL/AH/AHL); light and/or heavy chain deposition disease, light chain proximal tubulopathy (with and without crystals), GN associated with monoclonal immunoglobulin (with proliferative or membranoproliferative pattern), monoclonal fibrillary glomerulopathy, immunotactoid glomerulopathy, cryoglobulinemic GN (types I and II), crystalglobulinemia, crystal-storing histiocytosis, membranous nephropathy secondary to monoclonal immunoglobulin, renal infiltration by neoplastic cells; by indirect mechanism: C3 glomerulonephritis, thrombotic microangiopathy.
Here in this chapter we will review some of these entities, others are reviewed in the case discussion of our Case Series.
Light chain proximal tubulopathy - Case 80 of our Case Series. - - Another one: Case 114.
Light chain proximal tubulopathy with crystals - Case 179.
Proliferative glomerulonephritis with monoclonal Ig deposits - Case 91. - And another one: Case 142.
Fibrillary glomerulonephritis - Case 103.
Crystal-storing histiocytosis - Case 206.
Amyloidosis is defined by pathologic accumulation of extracellular proteins that adopt a beta-pleated configuration and share histochemical characteristics and fibrillary ultrastructure. The amyloidoses are a heterogeneous group of disorders with deposition of abnormally folded proteins in tissues. Amyloid deposits are formed from globular, soluble proteins, which undergo misfolding and, subsequently, aggregate into insoluble fibrils, leading to progressive organ damage. By electron microscopy, amyloid fibrils appear rigid, are non-branching, and typically measure 8–12 nm in diameter. These proteins are resistant to degradation. These substances share morphologic, ultrastructural, and staining features, but they have different chemical structure. Approximately 95% of the material that forms the amyloid consists of fibrillary proteins and 5% is P component and others glycoproteins.
The current classification of amyloid in medical practice is based on the amyloid protein type. The amyloid is termed A (for amyloid) followed by an abbreviation of the protein type: AL (amyloid derived from immunoglobulin light chain), ATTR (amyloid derived from transthyretin), etc. Amyloid protein variants associated with hereditary amyloidoses are named according to the substitution or deletion in the mature protein; thus, the name of the amino acid involved and the position of the change are listed: e.g., ATTRV30M (valine is replaced by methionine). While the single-letter amino acid code is recommended by the ISA, frequently, the three-letter amino acid designation is used in the literature, in keeping with the recommendations of the Sequence Variant Description Working Group of the Human Genome Variation Society. The name “hereditary” rather than “familial” is recommended by the ISA for amyloid diseases associated with mutant proteins. Moreover, “hereditary ATTRv,” where “v” stands for variant instead of “m” for mutant, is recommended by the ISA; however, at times, ATTRm (or hATTR for hereditary ATTR) may be encountered in the literature as an alternative to ATTRv. The designation ATTRwt is frequently used to underscore the association with wild-type protein in contrast to the variant (Picken MM. The Pathology of Amyloidosis in Classification: A Review. Acta Haematol. 2020;143(4):322-334. [PubMed link]).
The three main biochemical forms are:
1.) AL Protein: formed by light chains, mainly of lambda type, or by parts of them. This amyloid is the associated to monoclonal proliferation of B cells or plasma cells; when proliferation of these cells is not identified we call the disease “primary amyloidosis”. Although “primary amyloidosis” is a term used to denominate amyloidosis associated to B o plasma cell proliferations too, is better call it “amyloidosis associated to” (or “secondary to”) B o plasma cell proliferation.
2.) AA Protein: It is derived from a serum precursor denominated: SAA (serum amyloid-associated), a protein synthesized in the liver and that circulates united to HDL lipoproteins; it is an acute phase reactant present in elevated levels in the serum of patients with chronic inflammatory conditions such as tuberculosis, syphilis, osteomyelitis, rheumatoid arthritis, chronic inflammatory bowel disease, and others. Then, it is the amyloid deposited in amyloidosis secondary to chronic inflammations.
3.) Beta Amyloid: It is the amyloid found in cerebral lesions in Alzheimer’s disease. In a recent work, we demonstrated extracerebral small deposits in patients with familial Alzheimer’s disease (manuscript in preparation).
Others proteins that produce deposits of amyloid are: Transthyretin (prealbumin): in some types of genetic disease and senile systemic amyloidosis; Beta2-microglobulin (component of molecules of the major histocompatibility complex): amyloidosis associated to chronic hemodialysis. Other proteins produce located amyloidosis.
The amyloid term comes from “starch-like” and refers to the fact that tissues with amyloid turn blue after treatment with iodine followed by sulfuric acid, as starch does.
Until 2020, 36 proteins have been identified as being amyloidogenic in humans. Amyloid deposits may be localized or systemic, with deposits being present in a single organ in the former case and affecting various organs and tissues throughout the body in the latter. Exclusively localized amyloid deposits have been associated with at least 19 protein types, while at least 14 protein types (and many more variants) appear to be consistently associated with systemic amyloidosis. Interestingly, however, at least 3 protein types (most notably AL/AH, amyloidosis derived from immunoglobulin light or heavy chain, respectively, ATTR, and amyloidosis derived from β2 microglobulin, Aβ2M) can occur as either localized or systemic deposits (Picken MM. The Pathology of Amyloidosis in Classification: A Review. Acta Haematol. 2020;143(4):322-334. [PubMed link]). Systemic amyloidosis, with deposits of amyloid in internal organs, walls of sanguineous vessels, and connective tissues, is usually fatal and causes near one of each thousand deaths in developed countries (Pepys MB. Amyloidosis. Annu Rev Med. 2006;57:223-41. [PubMed link]).
Classification of amyloidosis tends to be based on the type of deposited protein, more than in the clinical characteristics: AL amyloidosis: associated to myeloma or other lymphoproliferative diseases; AA amyloidosis (or secondary to chronic inflammations): in chronic inflammations, neoplasms, rheumatoid arthritis, familiar Mediterranean fever, and others; AF amyloidosis (familial): several types associated to muted transthyretin; AE amyloidosis (endocrine): in located forms, i.e. endocrine tumors (medullary thyroid carcinoma and pancreatic islet cell tumors); Beta2 microglobulin amyloidosis: in chronic hemodialysis. Approximately 90% of cases of systemic amyloidosis are AL type and of the rest many are AA type.
In near 33% of AL amyloidosis cases myeloma is found, in the other cases usually plasma cell dyscrasias are demonstrated; sometimes there are less than 10% of plasma cells in bone marrow, but these cases show expression of a predominant light chain, suggesting monoclonality.
Physiopathology of the disease is complex; the reason for which these proteins are deposited forming beta-pleated sheet configuration is the key to understand the development of the disease. Many of these proteins are normally in the serum, usually in values very increased in patients with amyloidosis; some other molecules, as the P component (a glycoprotein), are united to precursory proteins of the amyloid and facilitates their aggregation, deposition, and organization. The nature of the biochemical interactions between these substances and the reason so that they are deposited in tissues are not completely understood.
Amyloidosis has an estimated prevalence of 1 in 60.000 people; in autopsies is 0.8%. It cause nephrotic syndrome in approximately 5% of the cases, being rarer in children (Heptinstall's Pathology of the Kidney, 5º ed.; Lippincott-Raven, Philadelphia, 1998, p. 1.339). There is slight masculine predominance for primary amyloidosis. In great series of biopsies of native kidney in adult patients in USA amyloidosis was the histologic diagnosis in 2% of cases, most of AL type; in contrast, in developing countries and Mediterranean countries renal amyloidosis is more commonly of AA type (Fogo AB y Kashgarian M. Diagnostic Atlas of Renal Pathology. Elsevier, Philadephia, 2005; p. 122). The age of the patients reflects the underlying clinical condition; patients with AL amyloidosis are typically adult and those that have amyloidosis associated to familiar Mediterranean fever are young.
Clinical features: Amyloidosis can involve any organ, but the deposits with more clinical relevance are in kidneys, heart and liver. Other frequent locations are skin, tongue, peripheral nerves, gastrointestinal tract, and spleen. In AL and AA type usually there is systemic involvement and frequently affecting kidneys. Renal amyloidosis produces proteinuria (80%) in a variable degree, in around 35-50% in nephrotic range and in some cases massive (>10g/day). There is serum creatinine increase in until half of the cases. Sometimes there are urine concentration defects due to tubulointerstitial deposits of amyloid. Extrarenal manifestations include liver dysfunction, peripheral neuropathy, cardiac failure, arrhythmias, hepato-splenomegaly, and macroglossia. In forms secondary to chronic inflammation the associated disease usually is evident: infections, skin lesions, collagen diseases, chronic inflammatory bowel disease, non-hematolymphoid tumors, and so on. When we have a patient with proteinuria in nephrotic range and systemic manifestations we must think, among other options, in amyloidosis. Cardiac involvement is unusual in AA amyloidosis and frequent in AL amyloidosis.
The prognosis depends to a great extent on the type, the extension of the disease, and the produced chronic damage. In AL amyloidosis the mean survival is between 9 and 20 years. AA amyloidosis can revert completely if the primary cause disappears.
Laboratory findings: Proteinuria, usually without hematuria; other findings of the nephrotic syndrome can be detected. There is altered renal function in until half of patients. Serum complement is normal. Serum protein electrophoresis will demonstrate monoclonal protein in most cases of myeloma; nevertheless, it is not detected in almost half of patients with AL amyloidosis; in many of these cases a monoclonal protein will be detected by immunoelectrophoresis (specific to detect some proteins like immunoglobulins or light chains). In 2/3 of patients there is a monoclonal protein in urine. Bence Jones protein is usually not detected.
In neoplastic proliferations producing Igs or fractions of them, physicochemical characteristics of the monoclonal proteins are very important for the type of disease that develops: if there are light chains that go through the glomerular filtration barrier it produces myeloma kidney: cast nephropathy, but if these proteins are so great size that they do not go through this barrier or they have chemical properties that prevent it, will be more probable that they deposit in tissues, producing amyloidosis or light chain deposition disease. It is exceptional that appears in a same patient disease by paraprotein deposition (amyloidosis or light [or heavy] chain deposition disease) and cast nephropathy (see renal involvement in myeloma).
By ultrasound or in the macroscopic study kidneys are increased of size, pale, and with smooth surface. Cortico-medullary differentiation is diminished.
Histopathology
Glomeruli are affected in almost all the cases of AA amyloidosis and in less than half of AL amyloidosis. The aspect is similar in both diseases. Deposits can be nodular or irregular in mesangium. In some cases there are deposits predominantly in mesangium (usually in earlier phases) and in others they are equally extensive in mesangium and glomerular capillary walls; usually they are more prominent surrounding the vascular pole (Figures 1, 2 and 3). Distribution of the changes can be irregular in a glomerulus and among glomeruli, but in long-standing disease deposits are usually more homogenously distributed. When the deposits are ample in mesangium, without formation of nodules, and involving the entire glomerulus, they adopt a pattern called mesangial diffuse. In capillary walls they can be subendothelial and/or subepithelial. In this last location they tend to form spikes perpendicular to the GBM giving a membranous GN aspect (Figure 4); I have had the opportunity to see a pair of cases initially misinterpreted like this GN; a careful histologic evaluation of mesangium and arterioles will allow us to arrive to the correct diagnosis and to carry out Congo red stain to confirm it. In advanced lesions there is a global replace of the tuft, with glomerulosclerosis appearance; nevertheless these glomeruli appear greater and Congo red positive; long time after, these deposits tend to disappear leaving only sclerosis. Usually there is no cellular proliferation. In cases with little glomerular involvement diagnosis can be difficult and requires high suspicion to request Congo red and/or electron microscopy.
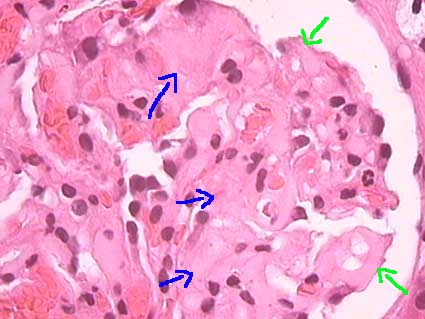
Figure 1. Amorphous, eosinophilic deposits in mesangium (blue arrows) and capillary walls (green arrows) that give a solidified aspect to the tuft (H&E, X400).
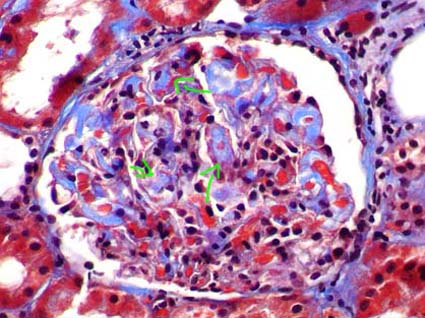
Figure 2. Amyloid deposits highlight with the trichrome staining (blue), like areas of mesangial acellular widening (arrows). (Masson’s trichrome, X400).
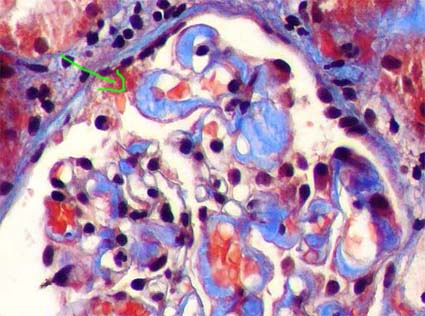
Figure 3. In some cases these deposits are more prominent in capillary walls, giving a thick and rigid aspect to them. The arrow indicates a capillary wall in which the deposits adopt a perpendicular disposition to this wall (see also Figure 4). (Masson’s trichrome, X400).
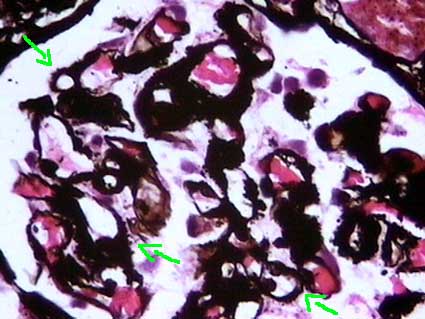
Figure 4. In cases with extensive capillary wall deposits we can see, with the silver stain, thick walls and spikes giving an aspect that can simulate membranous GN. Mesangial and extraglomerular deposits and the morphologic features with other stains must allow us to think in amyloidosis. (Methenamine-silver stain, X400).
Special stains are key for the diagnosis of amyloidosis; it is positive with Congo red, a stain easy to make and relatively specific; the true positividad is not the intense orange color, but the apple green birefringence under polarized light (Figures 5 and 6). In addition, there is dichroism: anisotropy in light absorption, it mean that the substance absorbs more light in one incident plane than another, so that light progressing through the material become more and more polarized as they proceed. This physical property causes that we see the green color at certain moment when crossing the lenses that generates the light polarization; collagen does not have this property, but it has (auto) refringence that is seen in any light incident plane, only changes its intensity. If the tissue is pre-treated with potassium permanganate (KMnO4) AA amyloid loses his affinity by the Congo red, in contrast, AL amyloid is resistant to this reagent; this technique can help to detect the type of amyloid.

Figure 5. Mesangial and parietal deposits in a case of primary systemic amyloidosis (AL). The Congo red stain shows reddish or intense orange deposits; nevertheless, the true positivity is the green birefringence (right) that presents dichroism. (Left: Congo red, X600; right: same field of the left seen with polarized light).
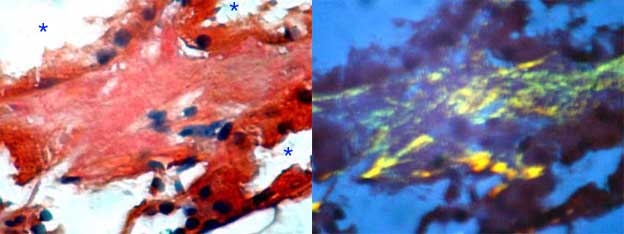
Figure 6. Interstitial deposits of amyloid. These deposits usually are irregular and focal, like the deposits in vessel walls. The asterisks indicate lumina of tubules in both sides of the interstitial zone with amyloid. (Left: Congo red, X600; right: same field of the left seen with polarized light).
Amyloid is eosinophilic (pink) with H&E; weakly PAS-positive (this fact assists in differentiating between amyloid and type IV collagen, and between amyloid and immune deposits, intensely positive); negative with methenamine-silver; it is metachromátic: purple with the violet crystal (unfortunately, relatively large amounts of amyloid must be present to demonstrate this property); and fluoresces green-yellowish with thioflavine T. By immunohistochemistry the P component can be detected (but it is unspecific, it is detected in all amyloid types) and AA amyloid can be confirmed with specific antibodies. Immunohistochemistry is not very successful to detect AL amyloid (there are many types of configuration of the molecule) or to detect kappa or lambda light chains.
Tubules show unspecific changes, although in some cases there is basement membrane thickening by deposits, more frequently in distal tubules and loops of Henle. Exceptionally the deposits can be predominant in tubules. In some cases there are intratubular cylinders that can show Congo red positivity. In interstitium irregular and focal amyloid deposits are usually detected, sometimes forming masses in the medulla. Vessel can have deposits of variable intensity and extension; and amyloidosis can involve arteries of all the sizes and veins; more frequently deposits affect arcuate and radiate arteries. In some cases, mainly in AL amyloidosis, arterial deposits can be predominant, with little or nothing of glomerular deposits.
Immunofluorescence
It is often difficult to interpret by entrapment of Igs and complement. Some works inform good correlation between their results for Kappa or Lambda chains, but in general, the results with this technique are complicated at the time of correlating with the type of light chain found in the serum or neoplasia (if any has been detected). Immunohistochemistry for light chains, in paraffin-embedded tissue, is still more difficult to interpret.
Electron microscopy
All the types of amyloid have a unique ultrastructural characteristic: liner, rigid-appearing, nonbranching fibrils. They have a diameter of 8 to 10 nm and may be up to 1 micrometer in length. They fibrils are randomly arranged and tend to form compact arrays when adjacent to cell membranes and more loose arrays when away from cell surfaces. When there are extensive deposits in the GBM, this is frequently fragmented and attenuated or may be completely absent in areas. In some cases GBM may have a double-contour with new material similar to GBM on the epithelial side of the deposits. Effacement of the epithelial foot processes may be evidenced.
[A revision in eMedicine© (link)]
See case 4: Primary systemic amyloidosis.
See Case 72: AA (secondary) amyloidosis.
Renal involvement in multiple myeloma (MM) can be due to obstruction and lesion of the tubules by light chains or fractions of them (myeloma kidney or cast nephropathy) or by deposits of paraproteins: amyloidosis and light chain deposition disease (LCDD). In this section we consider only myeloma kidney and in other sections of this chapter we deal with the other two.
MM present predominantly in the older population and affects more to men. It is the second lymphoproliferative disease more common in the general population and the first in Afro-Americans. Renal failure is the second cause of death in MM, and renal insufficiency is the most adverse prognostic indicator in patients with MM. Other complications of MM are: fractures, hypercalcemia, infections, and anemia. The lesions are predominantly on bones and produce small osteolitic zones with aspect in punch, or large with extensive bone destruction. Myeloma kidney (or cast nephropathy) is the most frequent cause of renal damage in MM, the second is amyloidosis (in 7-10% of patients with MM), and the third is LCDD (in approximately 5% of patients with MM).
In patients with MM there is anemia, back pain, bone fractures, serum creatinine increase in approximately 30% of cases, proteinuria in 70-80% of patients, and monoclonal spike (M component) in almost all the patients. In many cases IgG is synthesized complete, but in others only one light chain is synthesized which filters in the urine and can be difficult to detect in blood. Bence Jones protein (Ig light chains) is detected in urine of most patients and is very suggestive, but nonspecific, of MM (it can be detected in other conditions, including many causes of nephrotic syndrome).
Five years survival is approximately 18-27% in patients with MM, death usually is due to infection or renal insufficiency. The treatment is based on chemotherapy (frequently with melphalan) and prednisone. In addition, treatment of the dehydration, hypercalcemia and uric acid elevation are important issues. The nephropathy can revert in until 50% of patients.
Histopathology
The most relevant alterations in myeloma kidney are in tubules and interstitium. The casts occupy the distal part of the nephron: distal tubules and collecting tubules, but they are occasionally found in proximal convoluted tubules or even extending into Bowman’s space. They are variable in size with rigid and refractile aspect, strongly eosinophilic, weakly positive with the PAS stain and negative with methenamine-silver stain. With the processing and paraffin inclusion of the tissue, the casts may be seen separated of the epithelium and surrounded by a clear space. Cast may appear multilamellar and sometimes contain rhomboid or needle-like crystals. Some or many of the casts are surrounded by macrophages and multinucleated giant cells that migrate to tubules from the interstitium through ruptures of the basement membrane (Figures 7, 8, and 9). Sometimes tubular material can be seen in the interstitium due to basement membrane rupture. Neutrophils may be seen in tubules and casts. Casts contain Bence Jones protein and Tamm-Horsfall protein. The characteristic casts are not absolutely pathognomonic of MM without the appropriate clinical picture; similar casts can be seen in a number of other conditions (carcinomas, Waldenström macroglobulinemia…).

Figure 7. Tubular casts in myeloma kidney. Usually they are rigid and refractile. With trichrome stain they can be seen red or green (or blue according to the used technique). Many of them are accompanied by cells (see Figures 8 and 9). (Gomori’s trichrome, X400).
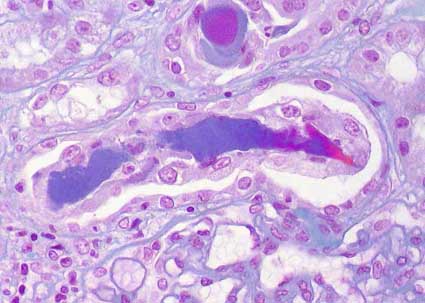
Figure 8. In this microphotography we can see two cylinders of amorphous material surrounded by multinucleated giant cells. This aspect is very suggestive of myeloma kidney (or cast nephropathy). (Masson’s trichrome, X400).

Figure 9. Fragmented cast in myeloma kidney surrounded by inflammatory cells that include neutrophils. Tubular epithelium is flattened, with atrophic aspect, and there are inflammatory cells. (H&E, X400).
The tubular epithelium is usually atrophic in tubules with and without casts (Figure 9); some appear expanded, but, if they do not contain cast are small and with thickened basement membrane. In some tubules epithelial regeneration is evidenced, which is correlated with the reversibility of the tubular injury in cases without atrophy.
The interstitium can have inflammation and fibrosis that correlates with the amount and size of the casts; they progressively increase of size until breaking and destroying the tubule. The extension of cast formation correlates with the tubular and interstitial damage and the severity of the alteration in the renal function.
In some cast there may be amyloid that can also be in small amounts in interstitium and vessels, but does not seem to have relevance in the renal damage. Usually there is no infiltration by neoplastic plasma cells in the kidney. Glomeruli are normal or with mild mesangial hypercellularity and/or basement membranes thickening. In vessels there are unspecific changes.
There is not cast nephropathy in all the patients with MM and the presence of abundant amount of Bence Jones protein is not always associated with this disease. There is no a direct relation between the severity of the nephropathy and the class of Ig, the type of light chain, the amount of Bence Jones proteinuria, or the uric acid level.
Similar cast may be seen Waldenström’s macroglobulinemia, acute renal insufficiency, thyroid carcinoma, and pancreas carcinoma, although in less quantity.
See myeloma cast nephropathy cases in our Case series: Case 5 -- Case 119 -- Case 132
[Very interesting Article: Herrera GA, et al., Renal pathologic spectrum in an autopsy series of patients with plasma cell dyscrasia. Arch Pathol Lab Med 128:875-9, 2004 (PubMed Link) (Free full text link])
Immunofluorescence
Glomeruli are negative for Igs and complement. In the cast any type of Ig can be identified, Kappa and Lambda light chains, albumin and fibrinogen. In some cases can be identified only the light chain produced by the monoclonal cells, but in other cases both chains can be identified. The immunostaining is characteristically more intense in the periphery of the cast and weak or negative in its center.
Electron microscopy
There are no specific ultrastructural alterations; the cast can be dense and homogenous or fibrillary. If there are crystals they are elongated, of varying size and, sometimes, with parallel arrays.
LIGHT (AND HEAVY) CHAINS DEPOSITION DISEASE
It is an infrequent complication of myeloma (MM) or other plasma cell dyscrasias. In many cases kappa light chains, or fragments of them, are deposited, but in other cases lambda is the light chain found (Kappa:lambda ratio: 4:1). The deposits are systemic, sharing many characteristics of amyloidosis; nevertheless, renal involvement is the one that dominates the clinical picture. Other organs involved are: heart, liver, lungs, skin, and endocrine glands.
Clinical features: The patients usually present renal failure, usually severe and relatively acute in onset. In most of patients there is severe nonselective proteinuria or nephrotic syndrome. The monoclonal protein is the predominant component in the urine in a few of patients. Hematuria can be detected in some patients. In some cases MM is not detected, only increase of plasma cells in bone marrow. In 90% of cases a monoclonal light chain is detected in the serum.
Histopathology
The most frequent finding in glomeruli is nodular glomerulopathy. Although there is variation in nodules size depending on the stage of the disease, these nodules often appear relatively homogeneous in size and they tend to be evenly distributed, whereas the nodules in diabetic nephropathy tend to be asymmetric. Mesangial nodules are composed of monotypic light chains and other proteins; they are PAS-positive but do not stain with silver stains (unlike nodules in diabetic nephropathy) and they are Congo red and crystal-violet-negative (unlike those in amyloidosis). Peripheral glomerular capillary walls are variably thickened (subendothelial deposition of light chains) and PAS-positive material can be identified. In some cases there is mesangial cellular proliferation; occasionally there is also endocapillary and/or extracapillary (crescents) proliferation (Figures 11 and 12).

Figure 10. See evident glomerular nodules, these have an acellular center with amorphous material that is negative with the silver and Congo red stains, which allows differentiating them of nodules in diabetic nephropathy and amyloidosis respectively. (Masson’s trichrome, X400).
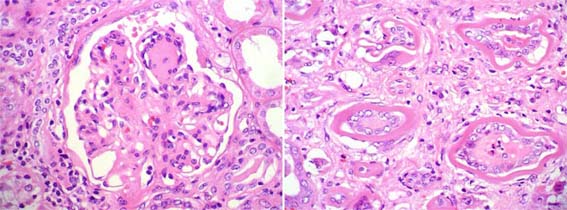
Figure 11. Nodular glomerulopathy. Note the ribbon-like material on the interstitial side of tubular basement membranes, with giant-cell reaction. (H&E, X400).
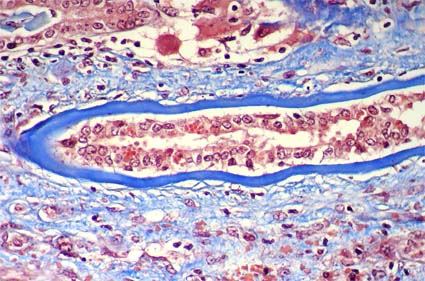
Figure 11b. tubular basement membranes appear thickened. This appearance is very useful in the diagnosis. (Masson's trichrome, X400).
Although the most striking lesion is in glomeruli, the most common finding is deposition of eosinophilic, refractil, ribbon-like material on the interstitial side of tubular basement membranes (TBM). This material is PAS-positive and appears brown on silver stain; TBM appear thickened and laminated. This change is more prominent in distal tubules, collecting tubules, and loops of Henle (search in deep cortex and medulla). Some tubules appear normal but deposits can be demonstrated with immunofluorescence. Tubular epithelium can be conserved, but it is frequent that shows some degree of atrophy. Similar deposits may be present within the walls of the arteries and arterioles, along with progressive dissolution of smooth muscle cells. The interstitium often shows fibrosis and PAS-positive deposits may be present. It is exceptional that coexists with cast nephropathy (myeloma kidney).
See Case 7 of our case series: Light chain deposition disease
Immunofluorescence
Usually there is linear staining of glomerular capillary walls for a light chain (usually kappa), with or without mesangial staining; nodules are also positive. There is strong, ribbon-like, linear staining around tubules, and there may be a diffuse staining in the interstitium. Deposits are more prominent in medulla and they may be absent in cortical tubules. Frequently there is positivity in Bowman’s capsule. In few cases there are deposits of both light chains, or deposits of a light chain and a heavy chain. When only a heavy chain is identified the disease is called: heavy chain deposition disease.
Electron microscopy
The peritubular deposits are granular and very electron-dense. In glomeruli they are identified in the nodules, mesangium, and subendothelials. In some cases there are not glomerular deposits, mainly in lambda light chain disease. These deposits may be also seen within the media of arteries and arterioles.
Less common forms of paraproteinemic disease in multiple myeloma include cryoglobulinemic glomerulonephritis and light chain proximal tubulopathy.
Light chain proximal tubulopathy is a form of proximal tubule injury secondary to increased filtration of pathogenic light chains. The classic and most widely recognized form of this disease has kappa-restricted intracytoplasmic crystals on renal biopsy and the clinical manifestation of a Fanconi’s syndrome. In recent years, the pathologic spectrum has been expanded to include cases with fibrillary cytoplasmic inclusions and those showing light chain restriction within the lysosomes without inclusions (Sanders PW. Mechanisms of Light Chain Injury along the Tubular Nephron. J Am Soc Nephrol. 2012 Sep 20. [Epub ahead of print] [PubMed link]; Herlitz LC, et al. Light chain proximal tubulopathy. Kidney Int. 2009;76(7):792-7. [PubMed link])
Proximal tubular damage by light chains results in acute tubular necrosis. The early changes include vacuolization of the tubular cells followed by apical blebbing with loss of surface microvillous borders, desquamation, and fragmentation. Monoclonal light chains may be detected in the cytoplasm of the tubular cell corresponding to the localization of the light chain in lysosomes (Heptinstall's Pathology of the Kidney, 6th ed. p. 864) (Figure 11c).
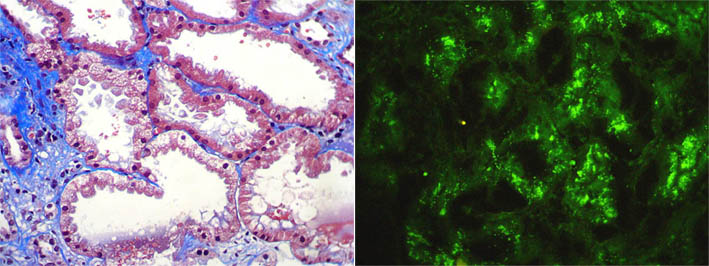
Figure 11c. Left: Acute tubular damage, with vacuolization of tubular proximal cells, blebbing and tubular dilatation. (Masson's trichrome, X400). Right: Direct immunofluorescence for kappa light chain, X400.
See Case 80 of our Case Series: Light chain proximal tubulopathy. --- See Case 114 of our Case Series. --- See Case 131 of our Case Series.
WALDENSTRÖM’S MACROGLOBULINEMIA
It is an uncommon B lymphocytes proliferative disorder characterized by the presence of a monoclonal serum IgM protein. This neoplasm can appear anywhere within hematolymphoid tissue and usually has the aspect of small lymphocytes or plasmacytoid cells. It is a low degree (indolent) lymphoproliferative disease that usually occurs in older patients. Symptoms are due to infiltration of the bone marrow or other organs by the neoplasm. Clinical presentation include anemia, elevated sedimentation rate, increased serum proteins, fatigue, weakness, recurring bleeding, lymphadenopathy, splenomegaly, high serum viscosity, and decreased fibrinogen. When the serum level of IgM is high (usually >3.0 mg/dL) appears hyperviscosity, cryoglobulinemia, hemorrhagic diathesis and peripheral neuropathy. Unusually there are osteolytic lesions or bone fractures. Occasionally there is amyloidosis.
Renal manifestations occur infrequently in this disease. Renal function is impaired in near 15% of patients with this condition. Acute renal failure is a result of extensive vascular occlusion by circulating IgM, dehydratation, massive renal infiltration by neoplastic cells, and distal nephron obstruction, as seen in myeloma. Occasional patients develop nephrotic or nephritic syndrome. Low excretion of Bence-Jones is frequent.
Treatment may be not needed until years after the diagnosis; treatment is usually required because of hematologic complications such as severe anemia, hyperviscosity, or visceral involvement. Oral alkylating agents are employed most often, with chlorambucil on a daily basis at low dose or intermittently at higher doses. The mean survival for all patients is 5 years. The death cause usually is due to the lymphoid malignancy, gastrointestinal bleeding and, occasionally, acute renal insufficiency.
The most frequent glomerular changes are variable sized subendothelial deposits; in some cases, well defined thrombi occlude capillary lumina. These deposits are PAS-positive and appear amorphous. Glomerular necrosis, which may be segmental, is rare and is a result of complete occlusion of glomerular capillaries or arterioles. Usually there is not hypercellularity. There are morphologic overlap between cases with Waldenström’s macroglobulinemia and those with cryoglobulinemia associated with other disorders. Amyloid can be seen in any of the renal compartments. Other reported lesions include minimal change disease, membranous glomerulonephritis, and cast nephropathy. Infiltration of the kidney by neoplastic cells can be seen in the disease. By electron microscopy capillary deposits and “thrombi” are electron-dense, amorphous and not fibrillary. These lesions are not identified in all the patients with Waldenström’s macroglobulinemia and they are not always associated with functional abnormalities.
Tubulointerstitial changes are unspecific.
MONOCLONAL GAMMOPATHY OF UNDETERMINED SIGNIFICANCE
Also known as benign monoclonal gammopathy and essential monoclonal gammopathy. It is defined by the presence of a monoclonal immunoglobulin in the plasma in absence of signs of B cells or plasma cells neoplasm. There must be less than 3g/dL of M protein and fewer than 5% of plasma cells in the bone marrow. Patients with systemic or renal disease related to paraprotein deposition are excluded of this category. Near 33% of patients will present multiple myeloma, lymphoma, or macroglobulinemia. Renal involvement is unusual and when appears usually it is due to proliferative GN, nephrosclerosis, focal and segmental glomerulosclerosis, and other glomerulopathies. In some cases amyloidosis, cryoglobulinemia, light chain deposition disease, or macroglobulinemia are documented, nevertheless, in these cases there is controversy with respect to the classification of the disease; most authors reserve the diagnosis “benign monoclonal gammopathy” for cases without these complications that implicate a worse prognosis.
FIBRILLARY AND IMMUNOTACTOID GLOMERULOPATHY
They are two diseases described relatively few years ago. They characterize by glomerular deposits of fibrils or microtubules. It is not clear if they are two different diseases or if they are variants of a same disease with different ultrastructural appearance. It seems better to separate them to know more about its etiopathogenic, clinical, and prognosis implications. Sum of the two diseases responds for approximately 4% of diagnoses in biopsies of patients with nephrotic syndrome (Korbet et al, Am J kidney Dis 27:647,1996 PubMed link) Fibrillary glomerulopathy has been reported as fibrillary glomerulonephritis.
Fibrillary Glomerulonephritis (FGN)
It is a disease with unknown cause, but it seems to originate by deposits of immune complexes. Igs in the deposits are polyclonal in most of cases (90%), with a minority (10%) showing monoclonality (only one light chain). Most of cases show a predominance of IgG subclass 4 (IgG4), which suggests some special property of this molecule that originate its particular ultrastructural arrangement. The disease presents with glomerular alterations, nonglomerular renal deposits and systemic involvement with organ dysfunction are very unusual (some case reports of pulmonary hepatic and cardiac involvement).
The mean age at presentation is 50 years with an equal distribution between men and women. It usually presents with nephrotic range proteinuria. Microhematuria is frequent. More than half of patients have nephrotic syndrome and all have proteinuria. Some patients are frankly nephritic or develop rapidly progressive glomerulonephritis. Many patients have renal insufficiency and hypertension at the diagnosis. Immunocomplexes, monoclonal proteins, and complement alterations are not detected.
Approximately half of cases evolve to terminal renal failure in few years (mean time: 2 years); the prognosis is worse in older patients or patients with renal insufficiency at diagnosis. The disease recurs post-transplantation in approximately half of the cases.
Histopathology
Glomerular alterations are very variable; a pattern of membranous GN, membranoproliferative GN, mesangial proliferative GN, or crescentic GN may be found. The most frequent changes include mesangial matrix expansion, thickening of capillary walls and mesangial hypercellularity. There are PAS-positive deposits in mesangium and capillary walls in many of the cases. With silver stains it is common to see alterations of the capillary walls: irregular aspect, subepithelial projections (spikes) and zones with empty aspect or holes. The deposits are negative with Congo red.
In interstitium, tubules, and vessels there are unspecific changes.
Immunofluorescence
There is IgG immunostaining in almost all the cases, usually accompanied by C3; with less frequency IgA and IgM are also identified. The staining is mesangial and parietal. In mesangium the deposits are confluent, and in capillary walls they can be irregular and granular, or ribbon-like, given a linear appearance. In most cases there is expression of both light chains, but in few cases (near 10%) there is only one light chain, usually Kappa (monoclonal). In tubules, vessels, and interstitium immune deposits are not detected.
Electron microscopy
The key ultrastructural feature is nonbranching, linear, randomly arranged fibrils resembling those of amyloid, but approximately twice as thick: 20 nm (range: 10 – 30 nm). They are located in mesangium, capillary walls (subendothelials, subepithelial, and, sometimes, inside GBM).The fibrils do not have an inner core or an organized structure. Electron-dense deposits may be present admixed with fibrils. There is effacement of podocyte foot processes and there is microvillous transformation of these cells, both findings being consequences of proteinuria. There may be also fibrillary deposits in Bowman’s capsule, but there are not in tubules or vessels.
See Case 103 of our Case Series: Fibrillary GN
Inmunotactoide Glomerulopathy (ITG)
It is a poorly understood disorder characterized by glomerular deposits with organized microtubules that probably represent immune complexes. In a report in 1980 Schwartz and Lewis coined the term immunotactoid glomerulopathy by analogy to the linear crystallization of hemoglobin S that forms elongated tactoids in red blood cells during sickle cell crisis (Schwartz MM, Lewis EJ. The quarterly case: nephrotic syndrome in a middle-aged man. Ultrastruct Pathol. 19801:575-82 (PubMed link)).The microtubules are larger than fibrils in fibrillary GN, almost always >30 nm (average 40 nm), although there is overlap between the two conditions. Microtubules have a substructure with a hollow core (or longitudinal central hollow), frequently adopt a parallel array, in contrast to the linear, randomly arranged fibrils in fibrillary GN. Occasionally there are microtubules with as small diameter as 20 nm. Although the diameter between cases is variable, usually is very homogenous in each case.
On immunofluorescence only a single light chain is detected (usually kappa) in the majority of cases, suggesting the presence of a monoclonal gammopathy. The deposits resemble that of the deposits seen in cryoglobulinemias and those in systemic lupus erythematosus.
Some cases have been associated with lymphoproliferative diseases and other systemic inflammatory diseases.
The clinical presentation is very similar to presentation in fibrillary GN; some authors find that the mean age at diagnosis is near 10 years older than the average in patients with fibrillary GN (60 years) and that there is minor probability of evolution to terminal renal failure, but it is not a finding reported in all the works.
Histologic features are very similar to those found in fibrillary GN.
On immunofluorescence there are deposits of IgG, C3 and, occasionally, other Igs, with the same characteristics that in fibrillary GN. Many of the cases express monoclonality for light chains kappa. In near to 10% both light chains are detected.
As for fibrillary GN, effective treatment is not known. The prognosis seems similar that fibrillary GN in some series, but it is better in other series.
The cryoglobulins are immunoglobulins (Igs) that reversibly precipitate in cold temperatures. There are three types according to the Igs contained: Type I: Monoclonal Igs; usually found in patients with multiple myeloma, monoclonal gammopathy of undetermined significance (benign monoclonal gammopathy) and Waldenström’s macroglobulinemia; usually it does not affect the kidney. Type II: There is a monoclonal Ig, usually IgM, with reactivity against IgG, in other words, an anti-IgG rheumatoid factor. Type III: they consist of IgG and IgM polyclonal antibodies.
Type II and III are known like mixed cryoglobulinemias, and they constitute the two third parts of cryoglobulinemia cases; Types I and II are considered a type of systemic vasculitis. They are associated to rheumatologic (connective tissue), infectious, lymphoproliferative, and hepatobiliary diseases. An important percentage of cases are associated to Hepatitis C virus (HCV). When there is not a known etiologic factor is named essential mixed cryoglobulinemia.
In most of cases renal disease is associated to type II cryoglobulinemia; it seldom occurs with type III.
Clinical features: The most frequent manifestations include cutaneous purpura and urticaria; in many cases there are weakness and arthralgias. Skin biopsy demonstrates leukocytoclastic vasculitis of small vessels. There is renal involvement in a variable percentage of cases (10-60%). The renal disease is, in most of cases, a subsequent event to the cutaneous lesions. The commonest renal manifestation is asymptomatic proteinuria with microscopic hematuria, renal function is usually normal. In one quarter of patients there is nephritic syndrome, with macroscopic hematuria, proteinuria, hypertension, and increased serum creatinine. There is oliguric or anuric renal failure in <5% of patients. Hypertension is frequent and often with difficult control. Occasionally there is proteinuria in nephrotic range.
The course of mixed cryoglobulinemia is variable; glomerular lesions usually have a more benign course than idiopathic membranoproliferative GN. There is chronic renal failure in approximately 10% of cases; this is developed after several years with the disease. Treatment is based on immunosuppression: corticosteroids and/or cyclophosphamide or others. In cases of severe acute crises plasmapheresis could be useful.
Laboratory findings: There is hypocomplementemia in most of cases, but usually it is more marked for C4, with C3 slightly decreased or normal. More important for the diagnosis is the detection of cryoglobulins in serum, almost invariably IgG and IgM with kappa light chain. In many patients antibodies against HCV or RNA of the virus are detected.
Histopathology
The commonest lesion is glomerulonephritis (GN) with morphologic aspects similar to type I membranoproliferativa GN (MPGN). Glomerular hypercellularity is predominantly by leukocytes and monocytes (different to idiopathic MPGN in which hypercellularity is mainly by mesangial and endothelial cells). Capillary walls show similar characteristics to those of idiopathic MPGN: double contours, “tram-tracking” aspect, duplication of the GBM and cells interposition in the subendotelial space (mainly monocytes) (Figure 12). Usually there are nodular, eosinophilic, hyaline, homogeneous deposits occupying some glomerular capillary lumina: intraluminal thrombi; when they are seen they are very suggestive of the disease (Figure 13). Crescents are not frequent and if present it is usually mild and in few glomeruli. In few patients there is mesangial hypercellularity and sclerosis, often associated to severe proteinuria and progression to chronic renal damage.

Figure 12. Glomerulus with notorious increase of size, nodular aspect, and thickening of capillary walls. With the other stains a MPGN aspect was demonstrated. In addition, there are intraluminal thrombi (see Figure 13). Patient with type II mixed cryoglobulinemia. (Masson’s trichrome, X400).
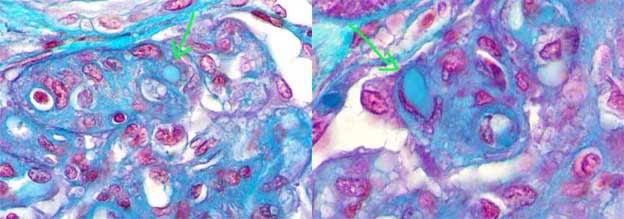
Figure 13. Same case of the Figure 12. The arrows indicate intracapillary hyaline thrombi; these are very characteristic of renal involvement in cryoglobulinemia. These “thrombi” are positive for immunoglobulins (in this case for IgG and IgM). (Masson’s trichrome, X600).
Cryoglobulins may be present in other systemic diseases as SLE, post-streptococcal GN, and others; GN in such patients may have histologic features resembling those of cryoglobulinemic GN and to be indistinguishable of it.
There are unspecific tubulointerstitial changes. In vessels we can find vasculitis of arterioles and small and medium size arteries in approximately one third of patients with cryoglobulinemic GN. There is fibrinoid necrosis of the wall that can be accompanied by neutrophils and monocytes. In some cases there are deposits similar to the glomerular hyaline thrombi in arterioles. In advanced stages we will see arterial chronic changes: intimal fibrosis.
Immunofluorescence
There are parietal subendothelial deposits of IgG and IgM, usually accompanied by C3 and C4, with a variable intensity. Intraluminal thrombi are strongly positive for IgG and IgM demonstrating their origin in immune complexes. Staining for IgG and IgM may be present in mesangium, arterial walls, and arterioles. When there is a monoclonal component (in type II cryoglobulinemia) the monoclonal origin can be demonstrated (usually kappa light chain).
See Case 161 of our Case Series: Mixed Cryoglobulinemia
See Case 194 of our Case Series: Lymphoma associated cryoglobulinemia
Electron microscopy
Glomerular lesions are very similar to those found in type I MPGN, but in cryoglobulinemia deposits are characteristic: organized, fibrillary or microtubular, with a distinctive substructure; there is a combination of curved cylinders with a width of 2.5 nm that are arranged in pairs and annular structures with spokes reaching 3 nm in diameter; they are in the subendothelial space and in the hyaline thrombi; occasionally they are found in the subepithelial space. The cells that interpose in the subendothelial space (between the original GBM and the new synthesized membrane, or between GBM and endothelial cell) correspond, in their majority, to monocytes; thus in cryoglobulinemic GN monocytes occupy the location that mesangial cells occupy in idiopathic MPGN.
Recent bibliography
Amyloidosis
Fibrillary and immunotactoid glomerulopathies
Cryoglobulinemias
Homepage - - - Tutorial Index