
Enfermedades vasculares
Hipertensión arterial, síndrome de Bartter, displasia fibromuscular de la arteria renal, embolismo de colesterol, necrosis cortical, síndrome hemolítico urémico/púrpura trombocitopénica trombótica, preeclampsia/eclampsia y vasculitis de medianos y grandes vasos
La filtración glomerular depende de un adecuado aporte sanguíneo a los capilares del penacho, por eso, las lesiones vasculares renales, además de amenazar la viabilidad del tejido, comprometen el equilibrio hidroelectrolítico y el mantenimiento del volumen corporal. Los riñones reciben el 25% del flujo cardíaco, una proporción más grande que cualquier otro órgano, por lo tanto las lesiones vasculares lo afectan de una manera muy significativa. Estas enfermedades suelen ser la manifestación de un compromiso vascular sistémico. Aquí nos centraremos en las alteraciones renales de origen vascular y dejaremos de lado las características del compromiso extrarrenal.
COMPROMISO RENAL EN HIPERTENSIÓN ARTERIAL SISTÉMICA
La hipertensión arterial sistémica (HTA) se ha clasificado de acuerdo con su etiología en esencial, primaria o idiopática (causa desconocida) y secundaria (causada por otras condiciones). También se ha clasificado como leve, moderada y severa, de acuerdo a las cifras de presión sistólica y diastólica. Algunas veces se utiliza el término hipertensión benigna para llamar a la hipertensión leve o moderada. La hipertensión maligna está definida por daño rápido (o agudo) de uno o varios órganos blanco (con papiledema), más que por una cifra de presión arterial. Inicialmente nos referiremos a los cambios renales de la hipertensión crónica (no maligna) y en un apartado diferente nos referiremos a los cambios renales en hipertensión maligna. A la afectación renal por HTA se le conoce también como nefropatía hipertensiva.
La HTA es un factor de riesgo para enfermedad renal crónica y de otros órganos, sin embargo, es usualmente asintomática hasta estados avanzados de lesión tisular. La fisiopatogenia de la HTA es compleja y extensamente tratada en otros textos. Aquí nos limitaremos a presentar las alteraciones renales secundarias a ella.
Cuando la enfermedad ha producido lesiones renales significativas, el órgano está reducido en tamaño y peso, la superficie capsular es usualmente granular y pueden verse depresiones secundarias a fibrosis cicatricial por isquemia, estos cambios son muy característicos de nefropatía hipertensiva. La corteza se adelgaza y frecuentemente hay quistes simples de tamaño variable.
Histopatología
Los cambios histológicos renales de la HTA crónica no son específicos. Las lesiones glomerulares y tubulointersticiales son de tipo isquémico y su aspecto microscópico es similar en todas las formas de lesión vascular con daño renal secundario a isquemia. Los glomérulos se retraen, las paredes capilares se ven gruesas y arrugadas, con aspecto ondulado o "en forma de acordeón" y hay un aumento relativo del espacio de Bowman; los cambios de la pared capilar se hacen más evidentes con las tinciones de PAS y plata (Figura 1). En algunos casos se evidencian lesiones esclerosantes segmentarias y focales y puden incluso asociarse a proteinuria (glomeruloesclerosis focal y segmentaria secundaria). Al avanzar el proceso, se comienza a depositar colágeno en la parte interna de la cápsula de Bowman (fibrosis intracapsular). Inicialmente el colágeno se deposita cerca del polo vascular y progresivamente va llenando toda la circunferencia del espacio de Bowman; el penacho queda comprimido en medio del tejido fibroso y el glomérulo termina en esclerosis global de tipo isquémico (Figura 2). En casos de esclerosis glomerular global el tejido fibroso y el penacho esclerosado tiñen con un aspecto similar con la H&E, pero con el PAS y la plata-metenamina se hace evidente el contraste entre el colágeno cicatricial que ocupa el espacio de Bowman (colágeno de tipo intersticial: fibrosis), que es débilmente positivo o negativo con ambas tinciones, y el colágeno de tipo glomerular en el penacho esclerosado (esclerosis), que es muy positivo con el PAS y con la plata. En muchos casos, con la plata-metenamina, se ven porciones o fragmentos de MBG en el centro del glomérulo esclerosado (Figura 2). Progresivamente, a lo largo de mucho tiempo, tiende a perderse el colágeno residual del penacho y, finalmente, sólo se ve tejido fibroso con desaparición del colágeno glomerular, haciendo, en algunos casos, difícil o imposible reconocer glomérulos esclerosados que se han mezclado con el tejido fibroso intersticial.
Figura 1. Glomérulo con cambios isquémicos iniciales; las paredes capilares se ven retraídas e irregulares, hay disminución del tamaño del penacho y un espacio de Bowman amplio, aún no hay esclerosis. Teóricamente estos cambios son reversibles. En muchos casos también identificaremos membranas basales tubulares con aspecto similar (no mostradas en la foto). (Plata-metenamina, X400).

Figura 2. Glomérulo globalmente esclerosado en el que se identifican fragmentos residuales de paredes capilares positivas con la tinción de plata (flecha); el material que ocupa el espacio de Bowman es colágeno de tipo intersticial, negativo con esta tinción. La tinción de PAS da un aspecto similar a estos glomérulos con esclerosis de tipo isquémica. (Plata-metenamina, X400).
Hay fibrosis intersticial y atrofia tubular en mútiples áreas, mezclándose con tejido conservado en el que se ven túbulos dilatados (dilatación compensatoria o "super túbulos"). En los túbulos atróficos hay disminución de su diámetro y de su luz, las células epiteliales se hacen bajas y a veces con citoplasma claro; el borde en cepillo de túbulos proximales desaparece. Las membranas basales tubulares se engrosan y pueden verse, al igual que en capilares glomerulares, irregulares, onduladas o arrugadas con el PAS y la plata. En el intersticio fibrótico, como en cualquier caso de fibrosis de otras causas, puede verse leve infiltrado inflamatorio mononuclear (Figuras 3 y 4).

Figura 3. Hipertensión arterial crónica de larga evolución en un paciente que falleció por enfermedad cerebrovascular. En el tejido renal podemos ver múltiples áreas de fibrosis, leve infiltrado inflamatorio mononuclear disperso, glomeruloesclerosis global focal (flechas azules) y fibrosis intimal en arterias (flechas verdes). (Tricrómico de Masson, X200).

Figura 4. Área cicatricial que adopta un patrón lineal desde la cápsula (derecha) la cual presenta una depresión (flecha). Este aspecto indica lesión obstructiva en la arteria que irrigaba esa zona ahora fibrótica. Si en la evaluación histológica de un riñón que va a ser trasplantado se tomara la muestra de esta zona, se sobrevaloraría el daño crónico y el porcentaje de glomeruloesclerosis; las biopsias de riñones donantes deben tomarse de zonas con aspecto normal macroscópicamente. (Tricrómico de Masson, X200).
En algunos casos hay hipertrofia del aparato yuxtaglomerular, sin embargo, esta hipertrofia es difícil de evaluar histológicamente cuando es en grado leve, necesitándose estudios de análisis de imágen (histomorfometría) para determinarlo con presición. Grados severos de hipertrofía son fáciles de identificar en la evaluación histológica con microscopía de luz convencional.
Arterias: hay engrosamiento concéntrico de la íntima por depósito de colágeno: fibrosis intimal; hiperplasia muscular de la media; disminución de la luz y síntesis de nuevas capas de lámina elástica: reduplicación de la elástica. La severidad de las lesiones renales se correlaciona con la severidad de la obstrucción luminal de arterias (Figuras 5, 6, y 7).
Arteriolas: El cambio característico son los depósitos hialinos en la pared de arteriolas aferentes: arterioloesclerosis hialina. Inicialmente se ven en el espacio subendotelial y progresivamente van aumentando de tamaño hasta ocupar o reemplazar la media muscular. Este material es muy positivo con el PAS, se ve homogéneo, eosinofílico, con la H&E y con el tricrómico puede verse rojizo o verde (o azul dependiendo del método utilizado). Los depósitos hialinos están irregularmente distribuidos, con segmentos libres de lesión y otros muy afectados (Figura 8). De acuerdo a la extensión de la arterioloesclerosis hialina en cada arteriola y a la cantidad de arteriolas comprometidas, la arterioloesclerosis hialina se ha dividido en tres grados: grado 1: pequeños depósitos en algunas arteriolas, grado 2: depósitos grandes en algunas arteriolas o pequeños en muchas arteriolas, y grado 3: extensos depósitos en la mayoría de arteriolas (o leve, moderada y severa). Recordemos que si identificamos arterioloesclerosis hialina comprometiendo tanto la arteriola aferente como la eferente debemos pensar en nefropatía diabética.

Figura 5. Arteria intrarrenal con leve fibrosis intimal; este cambio es muy frecuente en pacientes adultos, incluso sin diagnóstico de hipertensión. Los asteriscos señalan la capa muscular de la media; la zona interna, hacia la luz, muestra fibrosis leve (nefroangioesclerosis leve). (Tricrómico de Masson, X400).

Figura 6. Riñón con cambios vasculares moderados de hipertensión arterial. Hay fibrosis intersticial con leve infiltrado inflamatorio mononuclear, fibrosis intimal de arterias (flecha verde) y arterioloesclerosis hialina (flechas azules). (Tricrómico de Masson, X200).
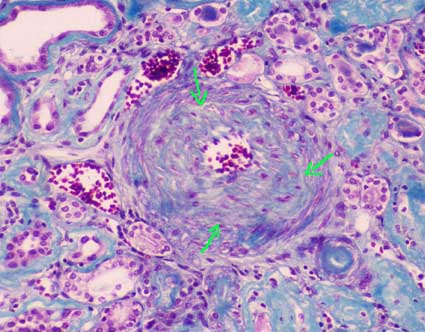
Figura 7. Arteria intrarrenal con severa fibrosis intimal que obstruye su luz en aproximadamente un 80%. Las flechas señalan el limite de la capa media muscular, todo lo que hay hacia su parte interna es proliferación fibrosa que obstruye la luz. (Tricrómico de Masson, X400).

Figura 8. Tejido renal de paciente con HTA sistémica de larga evolución. En arteriolas hay depósitos hialinos en su pared (flecha). La severidad de las lesiones arteriolares tienen algún grado de correlación con la afetación hipertensiva sistémica. (Tricrómico de Masson, X400).
Inmunofluorescencia
No hay depósitos específicos de Igs ni de complemento, pero en lesiones arteriolares hialinas y glomerulares segmentarias podemos ver depósito (o atrapamiento) inespecíficos de IgM y/o de C3.
Microscopía electrónica
No hay hallazgos específicos. Se evidencian las alteraciones de membranas basales: engrosamiento e irregularidad, y de las arteriolas: depósitos hialinos.
HIPERTENSIÓN MALIGNA
Los riñones pueden mostrar los cambios de HTA descritos antes si el paciente tiene historia de HTA de larga evolución, pero, cuando la enfermedad es de inicio reciente no encontraremos estos cambios crónicos. En la corteza y superficie suelen verse pequeñas hemorragias y petequias.
Histopatología
Las arterias presentan un notorio engrosamiento de la íntima por tejido conectivo laxo, de aspecto mixoide o mucoide, con células miointimales. La lámina elástica interna suele estar atenuada, sin reduplicación ni fragmentación. La luz está notoriamente disminuida. Pueden verse eritrocitos enteros o fragmentados en la íntima y algunas veces trombosis. Las arterias pequeñas y arteriolas presentan áreas con aspecto fibrinoide en su pared, desde la íntima hasta toda la media muscular: necrosis fibrinoide, y, con frecuencia, trombosis. La necrosis fibrinoide puede extenderse desde la arteriola hasta el glomérulo produciendo necrosis segmentaria o global del penacho. También es frecuente ver trombosis capilar intraglomerular (microangiopatía trombótica). Otras lesiones glomerulares que podemos encontrar son: mesangiolisis (marcada dilatación de capilares con mesangio apenas perceptible), formación de semilunas e hipertrofia del aparato yuxtaglomerular. Suele haber necrosis tubular aguda y, en ocasiones, pequeños infartos corticales (Figuras 9, 10 y 11).

Figura 9. Severa obstrucción de la luz de una arteria intrarrenal por proliferación de tejido laxo con células de núcleo grande, activo, en un paciente fallecido por daño cerebral agudo en hipertensión maligna. Con H&E el tejido intimal puede verse de aspecto mixoide. (Tricrómico de Masson, X400).

Figura 10. Arteriola renal del mismo caso de la microfotografía anterior. Observe el aspecto hialino en algunos segmentos de su pared, la presencia de material fibrinoide (rojo) en la íntima y eritrocitos fragmentados (flecha). Hay trombosis intraluminal. Lesiones similares pueden verse en microangiopatía trombótica de otras causas. (Tricrómico de Masson, X400).

Figura 11. En lesiones más tardías de HTA maligna hay fibrosis intimal concéntrica en arterias pequeñas y arteriolas (flecha verde). El glomérulo en esta imagen presenta esclerosis segmentaria (flecha azul) y el penacho no esclerosado muestra solidificación por aumento de matriz mesangial y disminución de luces capilares (flecha roja). (Tricrómico de Masson, X400).
En lesiones crónicas las arterias presentan engrosamiento intimal con aspecto concéntrico: en "piel (o bulbo) de cebolla" ("onionskin") (Figura 11) y cambios glomerulares y tubulointersticiales crónicos.
Las lesiones de la hipertensión maligna en riñón son similares a la que se ven en la escleroderma, que al fin y al cabo produce hipertensión maligna.
Por inmunofluorescencia se detecta fibrinógeno, IgM y C3 en las lesiones fibrinoides de arterias y arteriolas. Por microscopía electrónica se demuestra edema endotelial en arterias, arteriolas y capilares, con ensanchamiento del espacio subendotelial.
Es un síndrome caracterizado por hipopotasemia secundaria a la pérdida renal de potasio, alcalosis metabólica y una presión arterial normal o reducida. Suele haber hipomagnesemia e hipermagnesuria. Hay aumento de la síntesis de renina y aldosterona y, probablemente, de prostaglandina E2, prostaciclina, calicreína y bradicinina. La mayoría de casos se diagnostican en la infancia. Se presenta con poliuria y debilidad generalizada, atribuída a la hipopotasemia. El trastorno usualmente se hereda de manera autosómica recesiva, aunque se conocen casos esporádicos. El mecanismo fisiopatogénico no está completamente claro, pero parece que la clave está en una alteración en la reabsorción de cloruro en la rama ascendente gruesa del asa de Henle. La reabsorción inadecuada de cloruro determinaría una deplección de volumen y activaría el sistema renina-angiotensina. La liberación distal de sal y agua favorecería la secreción de potasio e hidrogeniones produciendo hipopotasemia y alcalosis. La síntesis de prostaglandinas aumentaría por la contracción de volumen y la hipopotasemia. Es posible que existan múltiples mecanismos fisiopatogénicos.
Un sistema ampliamente utilizado clasifica el síndrome de Bartter sobre la base de la genética subyacente, de la siguiente manera (tomado de: Frassetto LA. Bartter Syndrome, In eMedicine-Medscape: Ver el enlace):
Tipo 1 – Síndrome de Bartter prenatal: resulta de mutaciones en SLC12A1, el gen cotransportador de sodio-cloruro-potasio.
Tipo 2 – Síndrome de Bartter prenatal / neonatal: resultado de mutaciones en el gen ROMK.
Tipo 3 – Síndrome de Bartter clásico: causado por mutaciones del gen Kb del canal dependiente de voltaje del cloruro (CLCNKB), que codifica el canal de cloruro basolateral específico del riñón (ClC-Kb) involucrado en la reabsorción de cloruro de sodio en el túbulo renal.
Tipo 4 – Síndrome de Bartter con sordera neurosensorial: resulta de mutaciones de pérdida de función en BSND, que codifica una subunidad beta esencial para los canales de cloruro.
Tipo 5 – Síndrome de Gitelman: resulta de mutaciones en SLC12A3, el cotransportador de cloruro de sodio.
El diagnóstico no se basa en la histopatología, por lo que generalmente no se hace biopsia renal. El hallazgo renal característico es la hipertrofia difusa del aparato yuxtaglomerular y prominencia de células intersticiales medulares.
DISPLASIA FIBROMUSCULAR DE LA ARTERIA RENAL
Las causas de enfermedad renovascular son la ateroesclerosis, trombosis, compresión extrínseca y estenosis de cualquier causa. La enfermedad renal isquémica ocurre con una obstrucción superior al 70% de la luz. La displasia fibromuscular es una causa poco común de estenosis arterial, es más frecuente en mujeres y afecta predominantemente a adultos jóvenes. Se clasifica de acuerdo con la capa arterial comprometida:
- Fibroplasia intimal
- Hiperplasia medial
- Fibroplasia medial con aneurismas: la forma más común:
60-85% de los casos
- Disección medial: probablemente comienza en un área
de fibroplasia intimal
- Fibroplasia perimedial
La estenosis de la arteria renal principal o de sus ramas causa hipertensión renovascular por activación del sistema renina-angiotensina-aldosterona. La estenosis unilateral del riñón puede producir cambios de nefropatía hipertensiva en el riñón contralateral; el riñón con la estenosis está protegido de los efectos de la hipertensión; este fenómeno es conocido como "riñón de Goldblatt".
La displasia fibromuscular produce HTA y suele ser asintomática desde el punto de vista renal, hasta que se presentan lesiones avanzadas. Su tratamiento es quirúrgico y su pronóstico depende de la extensión de las lesiones y del daño crónico renal que se haya producido.
Histopatología
Las lesiones del parénquima renal son consecuencia de la isquemia debida a la lesión arterial y los cambios son similares a la isquemia de cualquier otra causa. Ver histopatología en "Compromiso Renal en Hipertensión Arterial Sistémica". Si la estenosis es unilateral, el riñón con lesión estenótica muestra fibrosis intersticial y atrofia tubular difusa; los glomérulos se ven más agrupados debido a la atrofia del parénquima cortical entre ellos; el aparato yuxtaglomerular puede estar hipetrofiado, y las arterias no suelen estar engrosadas ni mostrar cambios hipertensivos. El riñón contralateral (sin lesión estenótica) tiene esclerosis arterial y arteriolar y los demás cambios hipertensivos.
Fibroplasia Intimal: La íntima está engrosada por un tejido fibroso laxo, poco celular, la luz está reducida y la lámina elástica interna está conservada. Es una variante muy poco frecuente. A veces puede encontrarse acompañando otras formas de displasia fibromuscular.
Hiperplasia medial: Hay engrosamiento de la media con aumento del número de células musculares lisas. No hay cambios en la íntima, lámina elástica interna ni en adventicia. También es poco frecuente. Suele causar estenosis severa.
Fibroplasia medial con aneurismas: La forma más frecuente. Hay áreas de engrosamiento y fibrosis en la media que alternan con dilataciones aneurismáticas donde se pierde la lámina elástica interna. En las zonas de engrosamiento medial hay disminución de células musculares o aumento del espacio entre ellas. Es a menudo bilateral y usualmente no causa obstrucción completa de la arteria.
Disección medial: Hay defectos en la lámina elástica interna que permiten a la sangre llagar a la media y producir disección. Es probable que comience en áreas de fibroplasia intimal.
Fibroplasia perimedial: Hay acúmulos de colágeno reemplazando áreas grandes de la media muscular hacia la adventicia; la media está engrosada de una manera irregular.
Se presenta en pacientes con ateromatosis de la aorta abdominal o torácica o sus ramas; se suelen producir luego de procedimintos endovasculares o cirugia cardíaca. Con alguna frecuencia los vemos en injertos renales en los que el donante o el receptor presentan ateromatosis. También pueden producirse espontáneamente. Los anticoagulantes aumentan la probabilidad de desprendimiento de embolos de colesterol. Estos émbolos pueden ir a cualquier órgano produciendo complicaciones diversas de acuerdo al órgano y severidad de la obstrucción que produzcan. Los riñones son el órgano más comprometido.
Clínica: Suele presentarse varios días después del evento precipitante (usualmente cirugía); pueden verse lesiones en piel y en vasos de la retina (en el fondo de ojo). En riñón se manifiestan con elevación del BUN y la creatinina sérica. En el análisis de orina se detecta proteinuria, hematuria y cilíndros en algunos casos. La HTA puede ser importante. En muchos casos hay eosinofilia y eosinofiluria. El diagnóstico se puede demostrar en biopsias de piel. En muchos casos los émbolos son pocos, pequeños y asintomáticos. En casos de disfunción renal, algunos pacientes recuperan la función, pero en otros hay daño irreversible. No hay un tratamiento efectivo para este tromboembolismo.
Los riñones pueden mostrar infartos múltiples y lesiones en grandes vasos.
Histopatología
El hallazgo característico, y necesario para el diagnóstico, es la presencia de espacios vacíos con forma de aguja en la luz de arteriolas, arterias pequeñas y, ocasionalmente, arterias arciformes; estos espacios están rodeados por trombos de fibrina y, si han pasado varios días o semanas desde que se produjeron, células gigantes multinucleadas (Figura 12). Los espacios corresponden a los cristales de colesterol que se disuelven con el procesamiento normal del tejido. Para verlos es necesario hacer cortes por congelación de tejido fresco y mirarlos con luz polarizada. Émbolos pequeños pueden llegar hasta los capilares glomerulares y producir una reacción gigantocelular. En las primeras horas después del embolismo hay infiltrado de polimorfos que gradualmente se reemplazan por linfocitos e histiocitos. Dependiendo de la arteria comprometida podemos ver infartos de tamaño variable. Alrededor de embolos viejos se forma una reacción fibrosa.
Ver el Caso 188 de nuestra Serie de Casos: Embolismo de colesterol.

Figura 12. Arteria intracortical en la que hay distorsión de su aspecto debido a que la luz está ocupada por cristales de colesterol. Estos cristales desaparecen con el procesamiento del tejido y queda el espacio que ocupaban como agujas con extremos puntiagudos (romboidales elongados) (flechas verdes), se rodean de células inflamatorias, principalmente histiocitos gigantes multinucleados (flechas azules) y obstruyen parcial o completamente la luz. Los asteriscos señalan luz residual de la arteria. (Tricrómico de Masson, X300).
Es una causa poco común de falla renal en la que hay isquemia cortical severa con médula relativamente conservada. Se produce cuando hay falla circulatoria severa (choque) con o sin lesiones de arterias renales. Las causas más comunes son complicaciones obstétricas como abruptio de placenta, eclampsia y aborto séptico; síndrome hemolítico urémico; mordeduras de serpiente; hemólisis intravascular; rechazo severo del injerto, y colapso circulatorio de cualquier causa. La presentación clínica es la de una falla renal aguda. El compromiso es bilateral, aunque excepcionalmente puede ser unilateral.
Los riñones tienen una superficie oscura y con aspecto moteado, la corteza está pálida y la médula congestiva. Histológicamente la corteza presenta las características de un infarto isquémico que puede ser difuso o en parches. La corteza yuxtamedular, la médula y una pequeña banda de tejido cortical subcapsular (con irrigación de vasos de la cápsula) están conservados, con capilares congestivos. Pueden haber trombos en arterias y arteriolas. Con el tiempo las áreas necróticas tienden a calcificarse.
SÍNDROME HEMOLÍTICO-URÉMICO Y PÚRPURA TROMBOCITOPÉNICA TROMBÓTICA
El síndrome hemolítico urémico (SHU) y la púrpura trombocitopénica trombótica (PTT) son enfermedades estrechamente relacionadas y con manifestaciones clínicas muy similares, en las que es muchas veces imposible lograr una clara diferenciación. Las alteraciones renales son similares en ambas enfermedades. Las lesiones renales en arteriolas y capilares son similares en el SHU/PTT, eclampsia, hipertensión maligna, escleroderma, rechazo agudo humoral y toxicidad por ciclosporina, y se conocen como microangiopatía trombótica.
La lesión endotelial es clave en el desarrollo del SHU/PTT, el factor desencadenante de esta lesión puede ser endotoxinas bacterianas, anticuerpos contra células endoteliales, un factor inductor de apoptosis, ciclosporina, mitomicina, etcétera. Las células endoteliales alteradas actúan como procoagulantes y liberan factor de Von Willebrand facilitando la trombosis en pequeños vasos. También se han implicado en el desarrollo de la enfermedad una predisposición genética o deficiencia de algún antiagregante plaquetario. Se han descrito formas hereditarias de SHU y formas asociadas con anormalidades del complemento.
La infecciones que más frecuentemente causan o desencadenan SHU/PTT son E. coli, Shigella disenteriae, Streptococcus pneumoniae, Salmonella typhi y virus. Algunos casos se han asociado con embarazo, uso de anovulatorios orales, trasplante de médula ósea e irradiación al riñón. La mayoría de casos de PTT son considerados idiopáticos.
Clínica: El SHU/PTT es una coagulopatía de consumo con anemia microangiopática hemolítica, coombs negativo y trombocitopenia, sin otra causa aparente. Los pacientes presentan, además, fiebre, hipertensión arterial e insuficiencia renal. La enfermedad tiene predilección por el riñón y el sistema nervioso central; la PTT, especialmente por este último. La PTT ocurre principalmente en adultos y el SHU ocurre más en niños, principalmente las formas asociadas con diarrea. La insuficiencia renal es frecuente en ambas enfermedades y suele manifestarse por azoemia, hematuria macro o micro, proteinuria leve y cilindruria. En el SHU la afectación renal suele ser más severa y en la PTT suele afectarse, mucho más a menudo que en el SHU, el sistema nervioso central: alteraciones de la conciencia, signos de focalización, etcétera.
En la sangre de estos pacientes la prueba de coombs es negativa (descarta hemólisis autoinmune); los eritrocitos presentan cambios relacionados con la microangiopatía: fragmentación y esquistocitos. No hay alteracion en los niveles del complemento sérico. Para hacer el diagnóstico de SHU/PTT no es necesaria la biopsia renal.
El SHU ha sido dividido en dos variantes, una asociada a diarrea: síndrome hemolítico urémico asociado a E. coli productora de toxina Shiga (STEC-SHU) o SHU clásico o SHU diarrea positivo o SHU D+, y otra no asociada a diarrea: SHU atípico o SHU diarrea negativo o SHU D-.
STEC-SHU: Esta forma, también llamada forma clásica, se presenta luego de o durante un episodio de enfermedad diarreica, se presenta con falla renal aguda con oliguria, proteinuria, hematuria, anemia y trombocitopenia. Puede o no asociarse con compromiso sistémico. Podemos encontrar petequias en piel, púrpura, sangrado intestinal, hipertensión arterial y alteraciones neurológicas: convulsiones, afasia, problemas visuales, coma, etcétera. En estos casos la microangiopatía está, más a menudo, confinada a los glomérulos. Hay episodios relacionados con "brotes endémicos", principalmente en verano. El pronóstico es mejor en esta forma de SHU.
SHU atípico: puede presentarse en niños o adultos. No hay diarrea precediendo o durante el cuadro clínico. Hay falla renal e hipertensión más severas. La patogénesis del SHU atípico ha sido foco de mucha investigación y, hasta ahora, se ha asociado con la disregulación del complemento en hasta el 50% de los casos. Específicamente, mutaciones en proteínas reguladoras del complementio: factor H, factor I o factor B, o autoanticuerpos contra el factor H han sido implicados. Estas mutaciones resultan en la incapacidad para suprimir la activación del complemento y por razones que no se comprenden completamente, el endotelio glomerular es particularmente susceptible a estos cambios. El SHU atípico se ha asociado con infecciones no entéricas, virus, fármacos, tumores malignos, trasplante, el embarazo, y otras condiciones médicas subyacentes, tales como el síndrome antifosfolipído y esclerodermia. Las infecciones causadas por Streptococcus pneumoniae se han vinculado a 40% de casos de SHU atípico. Las categorías de medicamentos que han sido más frecuentemente asociados con SHU atípico incluyen moléculas contra el cáncer (mitomicina, cisplatino, bleomicina y gemcitabina), inmunoterapia (ciclosporina, tacrolimus, OKT3, IFN, y quinidina), y agentes antiplaquetarios (ticlopidina y clopidogrel). Al menos el 50% de los pacientes con SHU atípico tienen alguna anormalidad del complemento (hereditaria y/o adquirida), lo que conduce a una actividad disregulada de la vía alterna en la superficie de la célula endotelial. Sin embargo, existen otras alteraciones hereditarias no relacionadas con el complemento, como mutaciones en DGKE, que pueden resultar en alteraciones clínicas similares. El SHU atípico es una microangiopatía "trombótica" (MAT), cuyas características histopatológicas representan respuestas tisulares a la lesión endotelial. En algunas biopsias no se evidencian trombos (fibrina intraluminal o taponamiento por fibrina-plaquetas), sin embargo se encuentran alteraciones relacionadas con el daño endotelial: edema y desprendimiento de células endoteliales, mesangiólisis, dobles contornos de la membrana basal glomerular y acumulación subendotelial de material electrolúcido. En las arterias y arteriolas podemos encontrar fibrina intramural, engrosamiento intimal mixoide y proliferación miointimal concéntrica (en "piel de cebolla") (Goodship TH, et al. Atypical hemolytic uremic syndrome and C3 glomerulopathy: conclusions from a "Kidney Disease: Improving Global Outcomes" (KDIGO) Controversies Conference. Kidney Int. 2017;91(3):539-551. [PubMed link]).
Algunos clínicos ahora usan el término "SHU atípico primario" cuando se sospecha fuertemente una anormalidad subyacente de la vía alterna del complemento y se han excluido otras causas de SHU atípico. En muchos pacientes con un factor de riesgo subyacente en relación con el complemento, se requiere un desencadenante para que se manifieste el SHU. Los desencadenantes incluyen enfermedades autoinmunes, transplantes, embarazo, infecciones, fármacos y condiciones metabólicas. Puede ser difícil demostrar inequívocamente que un desencadenante desenmascare un defecto subyacente en la vía alterana del complemento. Tras el diagnóstico de una MAT se requiere una evaluación clínica y de laboratorio exhaustiva para establecer la etiología. Es urgente medir la actividad de ADAMTS13 para excluir púrpura trombocitopénica trombótica antes de decidir tratamiento con eculizumab y debería estudiarse la posibilidad de infección por E. coli. Idealmente, en todos los pacientes con sospecha de SHU atípíco dbería hacerse un estudio completo de la vía alterna del complemento (Goodship TH, et al. Atypical hemolytic uremic syndrome and C3 glomerulopathy: conclusions from a "Kidney Disease: Improving Global Outcomes" (KDIGO) Controversies Conference. Kidney Int. 2017;91(3):539-551. [PubMed link]).)
El pronóstico de la enfermedad es, en general, bueno. La mortalidad en la PTT es de aproximadamente el 20%; en el STEC-SHU es alrededor del 4% y en SHU atípico un poco más del 10%. En adultos el SHU es más heterogéneo en su presentación y la mortalidad es de hasta el 30%. Los casos de SHU en "brotes endémicos" tienen mejor pronóstico. La severidad de la hipertensión y la duración de la anuria son también factores relacionados con el pronóstico. En hasta el 39% de casos se encuentran cambios de nefropatía residual luego de SHU (Kern WF, Silva GF, et al, Atlas of Renal Pathology. W. B. Saunders Company, Philadelphia, 1999, p. 69). En su tratamiento se utilizan corticoides, antiplaquetarios, plasmaféresis, manejo de la insuficiencia renal y otras medidas de soporte. Eculizumab es el primer tratamiento aprobado por EE.UU. Food and Drug Administration (FDA) (septiembre de 2011) para los adultos y los niños con síndrome hemolítico urémico atípico.
Macroscópicamente los riñones presentan petequias subcapsulares; con frecuencia se ven áreas de necrosis. En casos de evolución prolongada los riñones disminuyen de tamaño y pueden presentar estrechamiento de luces arteriales.
Histopatología
Las paredes capilares de glomérulos están engrosadas debido al edema subendotelial, muchas veces dando un aspecto en doble contorno: por la separación del endotelio de la MBG o por neoformación de otra "MBG" entre la célula endotelial y la MBG verdadera; entre estas dos puede haber una células mesangial interpuesta. En la luz de capilares hay microtrombos de fibrina y plaquetas con eritrocitos atrapados y fragmentados. Estos trombos pueden llenar completamente el capilar o pueden adherirse a la pared sin ocluir por completo su luz. Cuando hay lesiones vasculares severas el glomérulo se ve distendido y con marcada congestión: "parálisis glomerular" y en casos aun más severos habrá franca necrosis del penacho. En ocasiones se ven acúmulos de polimorfos. El mesangio está ensanchado en algunos casos y con aspecto edematizado o apariencia fibrilar, sin hipercelularidad. Podemos ver también meangiolisis, caracterizada por pérdida de la matriz y células mesangiales con capilares que parecen sueltos y dilatados. En pocos casos se encuentra proliferación extracapilar (semilunas), usualmente pocas y pequeñas. En casos de lesiones vasculares severas podemos ver glomérulos con cambios isquémicos (ver alteraciones renales en HTA).
En lesiones avanzadas (o crónicas) vemos incremento de la matriz mesangial (esclerosis mesangial) con paredes capilares engrosadas y con doble contornos; o podemos ver cambios isquémicos. Los doble contornos pueden persistir por varios años, pero, en general, los glomérulos no presentan características similares a la GN membranoproliferativa ya que no suele haber hipercelularidad notoria (Figura 13). En casos de microangiopatía trombótica asociada a enfermedades por complejos inmunes, como lupus, encontraremos hipercelularidad glomerular y doble contornos que hacen confundir fácilmente con una GN membranoproliferativa tipo I idiopática.

Figura 13. Glomérulo de paciente con microangiopatía trombótica de varias semanas de evolución. En este glomérulo no se observan trombos, sin embargo, las paredes capilares están engrosadas por edema (flechas) con disminución de luces capilares, aspecto más "sólido" y sin hipercelularidad. Con plata-metenamina se observaron imágenes en doble contorno debido a edema subendotelial y formación de nuevo material similar a la MBG en su parte interna. (Tricrómico de Masson, X400).
En túbulos es frecuente ver cambios de NTA, cilíndros eritrocitarios y calcificaciones. En el intersticio hay edema y leve infiltrado inflamatorio mononuclear. En casos severos encontraremos áreas de necrosis y/o hemorragia.
Arterias: En algunos casos son normales; en estadios iniciales hay edema endotelial. Es frecuente encontrar necrosis fibrinoide de la pared arteriolar, sin células inflamatorias, acompañada de trombos arteriolares y eritrocitos fragmentados en la pared (Figura 10). Estos trombos pueden extenderse a los glomérulos y producir necrosis segmentaria o global. En algunas arteriolas hay dilatación (aneurismas), usualmente cerca del glomérulo; se dice que esta lesión es más común en PTT (Kern WF, Silva GF, et al, Atlas of Renal Pathology. W. B. Saunders Company, Philadelphia, 1999, p. 71). En casos de trombos organizados, el aspecto que toma la recanalización recuerda el penacho capilar, por lo que algunos autores llaman a estas lesiones trombóticas en organización: "estructuras glomeruloides". En lesiones de más tiempo de evolución la íntima está engrosada y con aumento de células (miointimales) y suele adoptar un patrón de engrosamiento concéntrico similar al visto en la hipertensión maligna: "en piel de cebolla"; de hecho, muchas de las alteraciones histológicas en el SHU/PTT son similares a las de la hipertensión maligna. La lesión residual es fibrosis intimal, difícil o imposible de distinguir de la de cualquier otra causa.
Inmunofluorescencia
Hay fibrina o fibrinógeno en paredes capilares glomerulares y lesiones arteriolares. También puede detactarse IgM, C3, IgG e IgA parietal en glomérulos. En arteriolas hay IgM y, en algunas ocasiones, C3, C1q y otras Igs. Estos depósitos de Igs y fracciones del complemento son inespecíficos, probablemente no patogénicos.
Microscopía electrónica
Hay edema del espacio subendotelial (en la lámina rara interna) y de las células endoteliales. Es frecuente ver interposición del citoplasma de células mesangiales entre la MBG y la célula endotelial. El espacio subendotelial es pálido, acelular y con apariencia fibrilar o granular. Hay áreas de desprendimiento del citoplasma de células endoteliales. En el mesangio la matriz tiene un aspecto finamente granular o fibrilar, algo similar a los cambios subendoteliales. Los procesos podocitarios se ven desprendidos de la MBG. Hay depósito de material similar al de la MBG en la parte interna, entre ésta y la célula endotelial o entre la célula mesangial interpuesta y la endotelial (es el material que origina el doble contorno). Además, se observan los trombos capilares y arteriolares, y las lesiones de la pared arteriolar como fueron descritos en histopatología.
Ver el Caso 97 de nuestra serie de casos
La preeclampsia está caracterizada por hipertensión, proteinuria y edema en el tercer trimestre del embarazo. La eclampsia está definida por la presencia de convulsiones en una paciente con los signos de la preeclampsia. Las características clínicas no son específicas y pueden presentarse en varias enfermedades renales o no renales subyacentes que se acentúan por los cambios fisiológicos del embarazo. La biopsia renal es muy pocas veces realizada en estas pacientes.
Los cambios histológicos son predominantemente glomerulares y se asemejan a los del SHU/PTT. Hay marcado edema e hipertrofia de células endoteliales: "endoteliosis". hay también vacuolización de células endoteliales y del mesangio, incremento de la matriz, usualmente, sin o con leve hipercelularidad mesangial, a veces, interposición mesangial e imágenes en doble contorno de la pared capilar (más frecuente en casos severos o en la fase de resolución). No es muy frecuente encontrar trombosis glomerular o arterial (Figuras 14 y 15). En arterias y arteriolas podemos ver cambios inespecíficos como fibrosis intimal, hipertrofia de la media y depósitos hialinos arteriolares; no suele haber necrosis fibrinoide.

Figura 14. Glomérulo de una paciente fallecida por eclampsia con síndrome HELLP; se evaluaron sus riñones por biopsia para posible trasplante; ambos fueron trasplantados y, a pesar de presentar retraso en el inicio de la función, luego de una semana empezaron a funcionar adecuadamente y siguen haciéndolo después de varios años post-trasplante. Observe este glomérulo con marcada disminución de luces capilares, paredes gruesas (flechas) y células endoteliales prominentes (endoteliosis). El glomérulo aparece sin elementos formes de la sangre (exangüe). En este caso hay leve hipercelularidad mesangial.(Tricrómico de Masson, X400).

Figura 15. Este otro glomérulo es del mismo caso de la microfotografía anterior. Hay un trombo capilar (flecha verde), hiperplasia e hipertrofia de podocitos, con mitosis en uno de ellos (flecha azul) y luces capilares deisminuidas. (Tricrómico de Masson, X400).
La inmunofluorescencia muestra hallazgos similares a los del SHU/PTT y en la microscopía electrónica podemos ver también, de acuerdo a la severidad de las lesiones, algunos hallazgos descritos en el SHU/PTT.
El pronóstico es excelente si recibe atención medica oportuna y adecuada. Los síntomas y signos resuelven rápidamente después de terminar el embarazo. Las lesiones histológicas desaparecen también en pocos días, usualmente sin secuelas morfológicas.
VASCULITIS DE MEDIANOS Y GRANDES VASOS
En el capítulo de glomerulonefritis extracapilar tratamos de las vasculitis de pequeños vasos que comprometen el riñón: granulomatosis de Wegener, poliangeitis microscópica y síndrome de Churg-Strauss. Además, en otros capítulos hablamos de la púrpura de Henoch-Schönlein y de las crioglobulinemias, que puede considerarse también otros tipos de vasculitis. Aquí nos referiremos a las vasculitis de vasos grandes y medianos. De ellas, la que más frecuentemente afecta el riñón es la poliarteritis nodosa, las restantes lo hacen muy raras veces.
Las vasculitis pueden definirse, de la manera más simple, como inflamación de la pared vascular. El riñón está afectado en muchas de ellas, algunas veces siendo el órgano más afectado y otras como una lesión leve que puede pasar desapercibida.
La nomenclatura más aceptada en vasculitis es la del Consenso Internacional de Chapell Hill de 2012, una versión revisada de la Conferencia de 1994: Jennette JC, et al. 2012 revised international chapel hill consensus conference nomenclature of vasculitides. Arthritis Rheum. 2013 Jan;65(1):1-11. [PubMed link]:
Vasculitis de grandes vasos
- Arteritis de Takayasu
- Arteritis de células gigantes
Vasculitis de medianos vasos
- Poliarteritis nodosa
- Enfermedad de Kawasaki
Vasculitis de pequeños vasos
- Vasculitis asociadas a ANCAs
- - Poliangiitis microscópica
- - Granulomatosis con poliangiitis (anteriormente granulomatosis de Wegener)
- - Granulomatosis eosinofílica con poliangiitis (Churg-Strauss)
- Vasculitis de pequeños vasos con complejos inmunes
- - Enfermedad anti-membrana basal glomerular
- - Vasculitis crioglobulinémica
- - Vasculitis IgA (Henoch-Schönlein)
- - Vasculitis urticarial hipocomplementémica (vasculitis anti-C1q)
Vasculitis de vasos de tamaño variable
- Enfermedad de Behcet
- Síndrome de Cogan
Vasculitis de un único órgano
- Angiitis cutánea leucocitoclástica
- Arteritis cutánea
- Vasculitis primaria del sistema nerviosos central
- Aortitis aislada
- Otras
Vasculitis asociada con enfermedad sistémica
- Vasculitis lúpica
- Vasculitis reumatoidea
- Vasculitis en sarcoidosis
- Otras
Vasculitis asociada con probable etiología
- Vasculitis crioglobilinémica asociada a Hepatitis C
- Vasculitis asciada a Hepatitis B
- Aortitis asociada a sífilis
- Vasculitis por complejos inmunes asociada a medicamentos
- Vasculitis asociada a ANCAs y medicamentos
- Vasculitis asociada a cáncer
- Otras
Otro abordaje para la clasificación es por su etiología (autoinmunidad humoral, pauciinmunes, infecciosas, mediadas por lesión celular). Es importante resaltar que, de acuerdo con la clasificación de Chapel-Hill, la presencia de lesiones de vasos pequeños es incompatible con el diagnóstico de vasculitis de vasos de mediano calibre, así algunas veces encontremos lesiones de arterias medianas en vasculitis de pequeños vasos. Y recordemos que los glomérulos son capilares, es decir, vasos de pequeño calibre. En otras palabras, si hay GN hay vasculitis de pequeños vasos.
Las vasculitis de pequeños vasos han sido tratadas en otros cápitulos (GN proliferativa extracapilar, Henoch-Schönlein y crioglobulinemias). Ahora revisaremos las lesiones renales en las vasculitis de vasos grandes y medianos.
Vasculitis de grandes vasos
En estas enfermedades raras veces hay compromiso renal. Pueden identificarse lesiones de la arteria renal principal y/o de arterias medianas intraparenquimatosas. La arteritis de células gigantes y la arteritis de Takayasu se caracterizan por inflamación granulomatosa de la pared arterial, más prominente en la capa media, aunque hay infiltrado linfo-histiocitario en toda la pared (panarteritis). En la fase activa hay granulomas, células gigantes y fragmentación de la elástica interna; ocasionalmente hay necrosis fibrinoide, pero en estos casos debemos pensar en otros tipos de vasculitis. La diferenciación entre los dos tipos de vasculitis de grandes vasos es más clínica que histológica. Hay informes de casos de arteritis de grandes vasos con compromiso glomerular, pero, de acuerdo con los conceptos actuales, debemos pensar que se tratan de dos enfermedades diferentes.
Vasculitis de vasos de mediano calibre
Poliarteritis nodosa (PAN): no la debemos confundir con la poliangeitis microscópica que es otra enfermedad clínica y anatomo-patológicamente diferente. Las lesiones vasculares se localizan en vasos de tamaño mediano: arterias interlobares, arciformes y corticales radiadas (interlobulillares). En la misma biopsia suelen coexistir lesiones en diferente estado evolutivo: agudas y crónicas. En fase activa hay necrosis fibrinoide en segmentos de las arterias, con infiltración de leucocitos; en fases tempranas predominan los neutrófilos que se fragmentan en su pared (leucocitoclasia), al avanzar el proceso van siendo reemplazados por linfocitos, monocitos y macrófagos. La necrosis inicia en la parte interna de la pared arterial y puede hacerse transmural, afectando tramos no continuos, por lo que a veces, puede no ser fácil encontrarla; con mayor frecuencia las lesiones se ubican en bifurcaciones. La necrosis fibrinoide se ve eosinofílica con H&E y roja con el tricrómico y se acompaña de cariorrexis y destrucción de la elástica interna (Figura 16). Podemos detectar trombos en zonas de necrosis, lo que puede originar infartos corticales que se ven triangulares. Los nódulos que le dan el nombre a la enfermedad corresponden a zonas de vasculitis con inflamación a su alrededor; se ven más frecuentemente en piel, pero pueden verse en otros órganos como el riñón. En algunos casos hay aneurismas por destrucción de la pared arterial (también originaría nódulos). En las fases crónicas de la lesión hay esclerosis vascular, recanalización de trombos y organización del tejido lesionado, todo esto puede llevar a hipertensión renovascular. En casos en los que sólo encontramos inflamación adventicial no podemos diagnosticar PAN, es necesario demostrar las lesiones agudas de la vasculitis. [Imagen de una arteria con necrosis fibrinoide de su pared (link)]
[Ver el Caso 108 de nuestra serie de casos]

Figura 16. Arteria cortical radiada con necrosis fibrinoide de su pared (flechas azules) y algunas células inflamatorias en medio de este material fuschinofílico (rojo). Las flechas verdes señalan segmentos de pared arterial sin lesión y los asteríscos la luz del vaso. (Tricrómico de Gomori, X400).

Figura 17. Arteria de mediano tamaño con necrosis fibrinoide de la pared, cariorrexis y trombosis que ocluye parcialmente su luz; este caso corresponde al de una paciente con poliarteritis nodosa. (H&E, X300).
Enfermedad de Kawasaki: También llamada "síndrome ganglionar mucocutáneo". Afecta predominantemente niños y se caracteriza por fiebre, linfadenopatía e inflamación de mucosas, piel y arterias: principalmente coronarias, axilares e ilíacas. Aproximadamente en un 25% hay compromiso de la arteria renal principal; raras veces se presenta disfunción renal.. Histológicamente, en arterias, hay edema, infiltración de la pared por neutrófilos y mononucleares, e inflamación perivascular. Al evolucionar el proceso disminuye el infiltrado de neutrófilos y persiste el linfo-histiocitario. Hay menos necrosis arterial que en la PAN; al final del proceso quedará fibrosis intimal variable. Ocasionalmente se describe compromiso de arterias intrarrenales.
Bibliografía
Nefropatía hipertensiva
SHU / PTT
Preeclampsia - Eclampsia
Vasculitis de medianos y grandes vasos