
Glomerulonefritis Proliferativa Extracapilar ("Crescentic")
Enfermedad anti-MBG - Glomerulonefritis pauciinmune
La glomerulonefritis proliferativa extracapilar (GNE) o con semilunas no es una enfermedad específica, sino, una manifestación histológica de daño glomerular severo. El nombre "extracapilar" o GN con "semilunas" (en inglés "crescentic") es utilizado para designar la proliferación celular y/o fibrosa que ocupa el espacio de Bowman, surgiendo desde su cápsula. Extracapilar se refiere a que está por fuera de los capilares del penacho glomerular. No hay acuerdo universal sobre el porcentaje de glomérulos que deben tener semilunas para llamarla GNE, algunos autores o textos hablan del 50%, otros del 30%, pero ese porcentaje de glomérulos con semilunas, para llamarla extracapilar, depende del contexto clínico. En GN asociadas a ANCAs es posible decir que es una GN extracapilar con sólo una semiluna. En GN mediadas por complejos inmunes hay más controversia, por ejemplo, en nefropatía IgA algunos autores proponen más del 50% y otros más del 30%. Más importante que eso, es cuantificar las semilunas en todos los casos; claro está, a mayor porcentaje de semilunas más agresivo se espera que sea el comportamiento clínico.
La definición de semilunas es la presencia de al menos dos capas de células que estén llenando total (circunferencial) o parcialmente (circunscrita) el espacio de Bowman.
Esta alteración es también conocida como GN rápidamente progresiva, GN subaguda y GN maligna (note que estos ultimos términos son clínicos, no histopatológicos). Se caracteriza histológicamente por semilunas y, clínicamente, por deterioro rápido de la función renal (semanas). Varias enfermedades glomerulares pueden producir este cuadro clinicopatológico y muchas de las glomerulopatías vistas en otros capítulos pueden producir semilunas. Sin embargo, en este capítulo veremos las enfermedades glomerulares en las cuales la GNE constituye la principal característica histológica.
Una GNE con clínica de GN rápidamente progresiva se puede ver en GN postinfecciosa, GN membranoproliferativa, nefropatía IgA, enfermedad de depósitos densos, etcétera, pero en estos casos es mejor llamarla de acuerdo a la glomerulopatía primaria, agregando el comentario "...con semilunas extensas en el X% de glomérulos".
Hay una clasificación de la GNE de acuerdo a las características inmunopatológicas:
Tipo I: Debida a anticuerpos anti-membrana basal glomerular (MBG).
Tipo II: Por complejos inmunes.
Tipo III: Sin depósitos de inmunoglobulinas o complemento en glomérulos: pauciinmune. Este grupo ha sido a su vez subdividido en: asociada a anticuerpos citoplasmáticos antineutrófilo (ANCAs) (poliangeitis microscópica, granulomatosis con poliangiitis [Wegener], granulomatosis eosinofílica con poliangiitis [Churg–Strauss], o vasculitis limitada al riñón) y no asociada a ANCAs (GNE idiopática).
Un cuarto tipo ha sido propuesto, por algunos autores, para aquellos casos en los que se documenta coexistencia de enfermedad anti-MBG y ANCAs. Estos tipos son claramente diferenciados en todos los textos de la materia, sin embargo, la denominación: tipo I, tipo II y tipo III se usa poco en el diagnóstico histopatológico.
En 2010 un grupo internacional de nefropatólogos propuso una nueva clasificación histopatológica basada en los hallazgos glomerulares:
Esquema de clasificación para la glomerulonefritis asociada a ANCAs (Berden AE, et al. Histopathologic classification of ANCA-associated glomerulonephritis. J Am Soc Nephrol. 2010;21:1628-36. [PubMed link] [Free full text]):
Focal: ≥50% de glomérulos normales.
Crescéntica: ≥50% de glomérulos con semilunas celulares.
Mixta: <50% normal, <50% semilunas celulares, <50% de glomérulos globalmente esclerosados.
Esclerótica: ≥50% de glomérulos globalmente esclerosados.
Esta clasificación es de ayuda para determinar un pronóstico al momento del diagnóstico y facilita un reporte uniforme entre centros.
La GNE mediada por complejos inmunes puede encontrarse en nefritis lúpica, púrpura de Henoch-Schönlein, nefropatía IgA, GN postinfecciosa, GN membranoproliferativa, crioglobulinemia, asociada a hepatitis B, a endocarditis y, en gerneral, a cualquier GN mediada por depósitos glomerulares de estos complejos. Cada una de estas enfermedades será tratada en su capítulo respectivo, en el actual presentaremos la enfermedad anti-MBG y la GNE pauciinmune.
La GNE corresponde a menos del 10% de todas las biopsias con diagnóstico de GN. De acuerdo a los tres tipos definidos la distribución es más o menos así: enfermedad anti-MBG: 20%; por complejos inmunes: 40%, y pauciinmune: 40%.
Origen de la proliferación extracapilar. Las semilunas están formadas por una proporción variable de células epiteliales y monocitos (macrófagos); de acuerdo a la evolución de la semiluna predominan los monocitos (fase inicial) o las células epiteliales (fases posteriores). También pueden identificarse otras células inflamatorias, principalmente linfocitos, y fibroblastos en fases tardías. En casos de GN con segmentos de necrosis del penacho y/o ruptura de la cápsula de Bowman suelen haber más monocitos, y en los casos de enfermedad por complejos inmunes hay más proporción de células epiteliales. Las semilunas están asociadas a ruptura de paredes capilares y fibrina en el espacio urinario. Esta fibrina y otras proteínas del plasma parecen tener la capacidad de estimular las células epiteliales para que proliferen, además, los mediadores liberados por monocitos y plaquetas contribuirían a la proliferación celular y, probablemente, la organización o fibrosis posterior. Las semilunas pueden evolucionar a fibrosis (semiluna fibrosa) o desaparecer (por apoptosis). La fibrosis es mediada por infiltración de fibroblastos desde el intersticio periglomerular a través de espacios en la cápsula de Bowman.
Esta enfermedad se presenta con frecuencia como un síndrome pulmón-riñón, caracterizado por hemoptisis, alteraciones pulmonares severas y una GN rápidamente progresiva (GNRP). Aunque desde hace muchas décadas se describieron pacientes con características clínicas similares, sólo en 1964 y 1965 se empezaron a describir depósitos lineales de gamaglobulinas en la MBG de dos pacientes con la enfermedad. Posteriormente fueron Lerner y cols. los que establecieron que los anticuerpos anti-MBG eran los causantes de las lesiones pulmonares y glomerulares (J Exp Med 126:989-1004, 1967 [Full text link]).
Desde la descripción inicial de Ernest Goodpasture de un paciente con enfermedad renal, pulmonar y vasculitis necrotizante esplénica e intestinal, ha cambiado mucho el entendimiento y nomenclatura de la enfermedad. El epónimo "Síndrome de Goodpasture" ha sido utilizado de manera variable en la literatura, pero actualmente se utiliza para denominar el síndrome pulmón-riñón producido por anticuerpos anti-MBG. Sin embargo, no todos los pacientes con anticuerpos anti-MBG presentan síndrome de Goodpasture: en algunos pacientes no hay afectación pulmonar.
Los anticuerpos anti-MBG están dirigidos contra la cadena alfa-3 en el dominio no colagenoso C-terminal del colágeno tipo IV (cadena alfa-3(IV) del dominio NC1). En pacientes con síndrome de Alport (sin formación de colágeno tipo IV de tipo adulto: sin cadenas alfa-3, alfa-4 ni alfa-5) no se desarrolla la enfermedad. El antígeno blanco está en riñón, pulmón, plejos coroides del ojo, cóclea. El anticuerpo reconoce el mismo antígeno en pacientes con S. de Goodpasture, GN aislada, o afectación pulmonar exclusiva, sin GN. Por lo tanto se considera que deben existir otros factores, adicionales a la presencia del anticuerpo, para que se desarrolle la enfermedad.
Se han identificado dos epítopes en el dominio NC1 alfa-3(IV) (EA y EB), ambos están ocultos entre el dominio NC1 y los protomeros de la triple hélice de colágeno. Por lo tanto, para que estos epítopes queden expuestos a los anticuerpos es necesario que haya una alteración que exponga estos antígenos. Al parecer, factores medioambientales, como el humo del tabaco o hidrocarburos, tendrían este efecto. También podría ser que oxidantes endógenos puedan abrir este sitio privilegiado, al igual que algunas subpoblaciones de anticuerpos anti-MBG (Hudson BG, et al, N Engl J Med; 348:2543-56, 2003 [PubMed link][Free full text]).
De una revisión de 2023: "Los estudios de mapeo de epítopos de α3(IV)NC1 han identificado varios epítopos nefritogénicos y residuos críticos que se unen a los autoanticuerpos y desencadenan la enfermedad anti-GBM. El descubrimiento de nuevos antígenos diana ha revelado la naturaleza heterogénea de esta enfermedad. Además, tanto la propagación como el mimetismo del epítopo se han implicado en la patogenia de la enfermedad anti-GBM. La propagación del epítopo se refiere al desarrollo de autoinmunidad a nuevos autoepítopos, lo que empeora la progresión de la enfermedad, mientras que el mimetismo del epítopo, que ocurre al compartir residuos críticos con péptidos microbianos, pueden iniciar la autoinmunidad. La comprensión de estas respuestas autoinmunes puede abrir oportunidades para explorar posibles nuevos enfoques terapéuticos para esta enfermedad. (Kuang H, Liu J, Jia XY, Cui Z, Zhao MH. Autoimmunity in Anti-Glomerular Basement Membrane Disease: A Review of Mechanisms and Prospects for Immunotherapy. Am J Kidney Dis. 2023;81(1):90-99. [PubMed link]).
Cuando los anticuerpos se ligan a la membrana basal activan complemento y proteasas, esta activación produce daño de la membrana y liberación de proteínas al espacio urinario facilitando la formación de semilunas. Linfocitos CD4 y CD8 inducen la migración de macrófagos y neutrófilos. La interleuquina 12 y el interferón gama contribuyen a la formación de la semiluna.
También hay factores genéticos relacionados con el desarrollo de la enfermedad. Personas con HLA-DRB1*1501 y DRB1*1502 tienen más suceptibilidad, mientras HLA-DR7 y DR1 son protectores (Kalluri R, et al, J Clin Invest 100:2263-2275, 1997 [PubMed link] [Free full text]).
La enfermedad anti-MBG es poco frecuente, afecta a personas de cualquier edad, pero es más común en pacientes de 20 a 40 años. Hay mayor frecuencia en hombres que en mujeres.
Clínica: Usualmente se manifiesta como una GNRP, a menudo acompañada por hemorragia pulmonar. Aunque muchos pacientes presentan enfermedad severa pulmonar y renal, el rango es amplio y en algunos casos puede haber sólo hematuria y proteinuria sin insuficiencia renal ni alteraciones pulmonares. En ocasiones la enfermedad es precedida por un episodio catarral, aunque se desconoce si una infección viral puede ser el desencadenante del cuadro clínico. Aproximadamente una tercera parte de los casos se acompaña de hemorragia pulmonar manifestada con hemoptisis, disnea y roncus, aunque en el seguimiento este porcentaje se incrementa hasta aproximadamente el 50%. La hemorragia pulmonar puede llevar a anemia importante.
La enfermedad renal se caracteriza por falla renal oligúrica o anúrica, hematuria macro o microscópica y otros síntomas generales inespecíficos. De acuerdo a la retención de sodio y la expansión del volúmen de líquidos puede haber hipertensión.
El tratamiento se basa en altas dosis de esteroides, otros inmunosupresores y plasmaféresis. La enfermedad anti-MBG tiene peor pronóstico que la GN por complejos inmune y que la pauciinmune. Alrededor de la mitad de los pacientes desarrolla falla renal terminal. Después de una remisión completa pueden haber episodios de recidiva aun años después del inicial. La enfermedad también puede recidivar post-trasplante, pero si no hay anticuerpos anti-MBG detectables el riesgo es pequeño.
Ver el Caso 93 de nuestra serie de casos: Enfermedad anti-MBG.
Histopatología, inmunofluorescencia y microscopía electrónica: Ver más adelante.
Glomerulonefritis extracapilar pauciinmune
GN extracapilar (GNE) sin depósitos bien definidos de inmunoglobulinas es llamada pauciinmune. La enfermedad puede hacer parte de un cuadro de vasculitis sistémica y, ya que afecta capilares glomerulares, se encajaría dentro de las vasculitis de pequeños vasos. Cuando no hace parte de una enfermedad sistémica y hay afectación sólo renal, se le conoce también con los nombres de GNE idiopática, GNE primaria o de vasculitis limitada al riñón.
Aproximadamente el 90% de pacientes con GNE pauciinmune son ANCA positivos. Son muy raros los casos de GNE idiopática, no asociada a ANCAs, complejos inmunes ni a anticuerpos anti-MBG (aproximadamente 5%). La GNE pauciinmune es la causa más frecuente de GN rápidamente progresiva y del síndrome pulmón-riñón. El diagnóstico diferencial en GNE pauciinmune es: granulomatosis de Wegener, poliangeitis microscópica, síndrome de Churg-Strauss y GNE idiopática.
El diagnóstico de granulomatosis con poliangeitis (Wegener) se basa en la demostración de vasculitis granulomatosa. Los granulomas se demuestran más frecuentemente en tracto respiratorio, son muy inusuales en tejido renal, pero, cuando se identifican en este órgano deben estar separados de glomérulos con ruptura de la cápsula de Bowman, ya que cuando esto ocurre es muy frecuente la formación de granulomas como una reacción a dicha destrucción. Diagnosticamos granulomatosis eosinofílica con poliangeitis cuando hay una historia de asma y eosinofilia y hay granulomas pulmonares; y diagnosticamos poliangeitis microscopica cuando hay una vasculitis sistémica sin granulomas ni historia de asma. Hay evidencia clínica o morfológica de afectación renal en el 90% de pacientes con poliangeitis microscópica, 80% de pacientes con G. de Wegener y 45% de pacientes con Churg-Strauss.
El papel de los ANCAs en la patogénesis de estas enfermedades no está completamente entendido. Los ANCAs pueden estar dirigidos contra mieloperoxidasa (P-ANCAs: patrón de tinción perinuclear) o contra proteinasa 3 (C-ANCAs: patrón de tinción citoplasmático). El patrón citoplasmático o perinuclear se refiere al aspecto que toma la tinción cuando se hace inmunofluorescencia indirecta en neutrófilos fijados en alcohol; el patrón perinuclear parece ser un artefacto por precipitación del sustrato. En Wegener usualmente hay C-ANCAs y en P. microscópica hay más frecuencia de P-ANCAs, y en pacientes con GNE limitada al riñón y pacientes con Churg-Strauss hay predominantemente P-ANCAs, aunque hay variabilidad.
Sabemos que los títulos de anticuerpos correlacionan con el grado de actividad de la enfermedad. Los ANCAs pueden activar los neutrófilos y producir su degranulación y liberación de radicales libres. El mecanismo de activación parece implicar unión a receptores de Fc y unión de Fab'2 en la superficie celular (Kettritz R, et al, J Am Soc Nephrol; 8:386, 1997 [PubMed link]). Los neutrófilos activados por ANCAs lesionan el endotelio causando daño de vasos (vasculitis); la necrosis endotelial parece ser un evento inicial en la patogénesis. De acuerdo con algunas hipótesis, en pacientes con ANCAs circulantes, un evento inflamatorio, como una infección respiratoria, prepara (o inicia) los neutrófilos y monocitos para ser activados por ANCAs; aproximadamente un 90% de pacientes relata un episodio catarral antes del inicio de los síntomas.
Clínica: Como en el caso de la enfermedad anti-MBG, usualmente se presenta con un cuadro de GN rápidamente progresiva. El inicio suele ser con oliguria, elevación de la creatinina, proteinuria variable, hematuria macro o micro, e hipertensión arterial. Ocasionalmente hay síndrome nefrótico. En algunos pacientes la creatinina sérica puede estabilizarse en niveles altos, pero en otros progresa hasta insuficiencia renal crónica terminal.
La afectación extrarrenal incluye fiebre, artralgias, mialgias, púrpura, neuropatía periférica, alteraciones respiratorias y signos de vasculitis en tracto digestivo. Hay lesiones cutáneas (púrpura y/o nódulos) en cerca de la mitad de casos. En Churg-Strauss hay afectación extrarrenal por vasculitis en un porcentaje alto de casos. En Wegener hay mayor compromiso de tracto respiratorio superior por lesiones granulomatosas necrotizantes o por vasculitis (80-90%). En el pulmón pueden evidenciarse nódulos (granulomas necrotizantes) y cavidades; son frecuentes la hemorragia alveolar, arteritis y capilaritis (afectación pulmonar en 90%). En P. microscópica hay compromiso de tracto respiratorio superior en una tercera parte de casos y compromiso pulmonar en la mitad. La presentación clinica de la G. de Wegener y de la P. microscópica no permite hacer un diagnóstico diferencial; sólo se hace con la demostración (o ausencia) de granulomas. En muchos casos sin estos últimos no se pueden distinguir con precisión estas dos enfermedades; sin embargo, la relevancia para el manejo no es muy grande dado que el tratamiento es similar.
Aproximadamente el 90% de pacientes responde a un tratamiento inmunosupresor agresivo (usualmente ciclofosfamida y esteroides) con tasas de remisión completa cercanas al 75%. Muchos pacientes presentan recaídas que responden de nuevo al tratamiento en un 70%. En algunos casos severos puede hacerse plasmaféresis pero hay controversia respecto a los resultados.
Datos de laboratorio: En la enfermedad anti-MBG y en la GNE pauciinmune podemos encontrar los mismos hallazgos en la orina y en los estudios de función renal: hematuria macro o microscópica, cilíndros eritrocitaros en el sedimento, proteinuria variable y elevación de BUN y creatinina. En la mayoría de pacientes con enfermedad anti-MBG se detectan los anticuerpos en suero, pero es una técnica difícil en la que puede haber falsos negativos. Se detectan ANCAs en aproximadamente el 80-90% de pacientes con Wegener y poliangiitis microscópica y en 60% de pacientes con Churg-Strauss. En aproximadamente una tercera parte de casos de enfermedad anti-MBG se detectan ANCAs, pero es difícil conocer su papel patogénico; estos pacientes parecen tener mejor pronóstico que aquellos con anticuerpos anti-MBG sin ANCAs. El complemento suele estar en niveles normales.
El aspecto histopatológico en el tejido renal es muy similar en la enfermedad anti-MBG y en las GNE pauciinmunes. Hay GN con lesiones necrotizantes focales o difusas y segmentarias o globales, lo más habitual es que sean segmentarias comprometiendo un número variable de glomérulos (Figura 5); las lesiones necrotizantes están invariablemente acompañadas de semilunas, aunque éstas también se ven en algunos glomérulos sin necrosis. Las semilunas se asocian a zonas de ruptura de las paredes capilares (más fáciles de evidenciar con la tinción de plata-metenamina) (Figura 2) y depósitos de fibrina en el espacio de Bowman; las semilunas están formadas por células epiteliales proliferadas y monocitos, a veces con linfocitos y polimorfos. Cuando no hay colágeno ni otros elementos de tejido fibroso se denomina semiluna epitelial (Figuras 1 y 2). Al avanzar el proceso migran fibroblastos que sintetizan colágeno que remplaza progresivamente la semiluna (más fácil de evidenciar con tricrómico o con plata-metenamina), cuando se mezclan componentes celulares y colágeno se denomina: semiluna fibroepitelial (Figura 3); en estados avanzados, sin proliferación celular y sólo matriz fibrosa y fibroblastos, se denomina: semiluna fibrosa (figura 4). Estos tres estados nos indican el grado de actividad o cronicidad y por lo tanto, en alguna medida, dan idea de la respuesta al tratamiento.
Figura 1. Paciente de 58 años de edad con GN rápidamente progresiva. Observe la proliferación de células grandes que ocupa todo el espacio de Bowman (flechas verdes) comprimiendo el penacho (flechas rojas). La cápsula de Bowman está señalada por las flechas azules. Este es el aspecto característico de una semiluna epitelial, en este caso circunferencial. Las células que forman la semiluna pueden ser epiteliales, monocitos u otras células inflamatorias. (Tricrómico de Masson, X400).
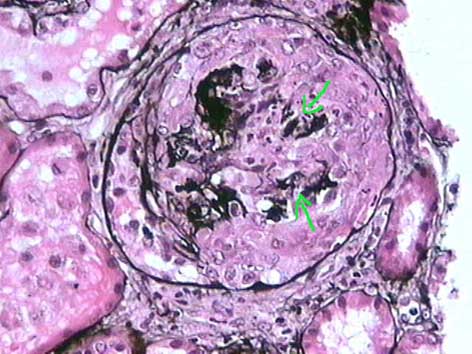
Figura 2. En las semilunas epiteliales las células que proliferan no se acompañan de tejido fibroso o colágeno. La plata resalta el penacho comprimido y con ruptura de paredes capilares (flechas), este daño capilar es un fenómeno patogénico importante en la generación de la proliferación extracapilar. (Plata-metenamina, X400).
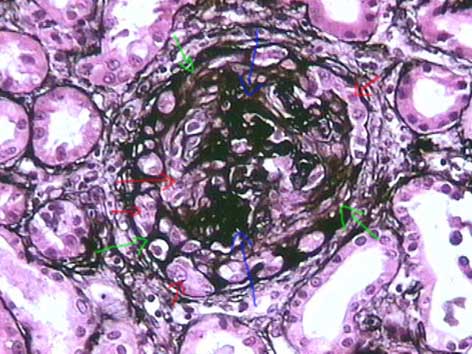
Figura 3. En las semilunas fibroepiteliales podemos ver una mezcla de células grandes, con núcleo redondeado, activo, muchas veces con nucléolo prominente y citoplasma amplio, que se mezclan con tejido fibroso o colágeno que resalta con la tinción de plata (como en esta foto) y con la tinción de tricrómico. Las flechas azules señalan el penacho comprimido y distorsionado, las rojas señalan células grandes, activas, que indican que aún hay proliferación epitelial, y las verdes la fibrosis que está reemplazando muchas áreas de esta semiluna, indicando un proceso que evoluciona a la cronicidad. (Plata-metenamina, X400).
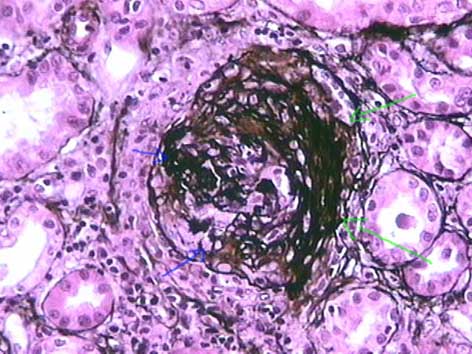
Figura 4. Al avanzar el proceso destructivo del glomérulo la semiluna se hace más fibrosa (o cicatricial) y las células que la componían disminuyen progresivamente hasta desaparecer, siendo reemplazadas por fibroblastos. Las tinciones de plata y tricrómico resaltan el componente fibrosos (flechas verdes), en este caso sin componente epitelial; las flechas azules señalan lo que queda del penacho glomerular. (Plata-metenamina, X400).
En el penacho, además de necrosis, puede identificarse colapso capilar, incremento de la matriz mesangial y cariorrexis. Con frecuencia hay destrucción segmentaria o extensa de la cápsula de Bowman (Figura 5); en estos casos es usual encontrar granulomas y células multinucleadas rodeando el glomérulo, estos granulomas no indican G. de Wegener.
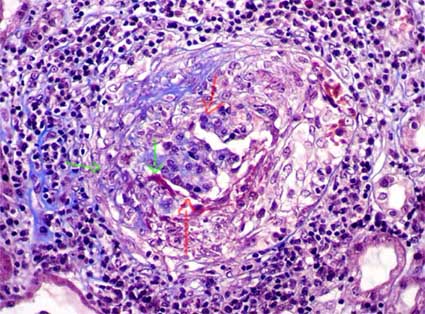
Figura 5. En este glomérulo podemos ver el penacho deformado y con un área de fibrina y fragmentación de núcleos (cariorrexis) (flechas verdes) que indican necrosis segmentaria del penacho. Hay una semiluna circunferencial epitelial, son un área evolucionando a fibroepitelial (parte superior izquierda de la circunferencia glomerular). Las flechas rojas señalan el penacho capilar comprimido. El infiltrado inflamatorio mononuclear es prominente alrededor de este glomérulo; este severo infiltrado se asocia con ruptura de la cápsula de Bowman. En algunos casos podremos evidenciar granulomas periglomerulares que no indican granulomatosis de Wegener. (Tricrómico de Masson, X400).
En vasos puede haber vasculitis transmural, con necrosis fibrinoide y/o infiltrado linfocítico y de polimorfos fragmentados (leucocitoclasia) (Figura 6). Cualquiera de los vasos puede estar comprometido, siendo las lesiones arteriales las más fáciles de evidenciar. En intersticio es usual encontrar notorio infiltrado inflamatorio mononuclear, edema y, en estados más avanzados, fibrosis. En túbulos pueden haber lesiones agudas: necrosis tubular, degeneración del epitelio y tubulitis, y lesiones crónicas (atrofia). En enfermedad anti-MBG los anticuerpos pueden también reconocer antígenos en la basal tubular presentando mayor daño tubular e inflamación intersticial. Los granulomas intersticiales no asociados a glomérulos nos indican que se trata de una G. de Wegener, pero son muy inusuales en riñón; habitualmente tienen un centro necrótico y estan rodeados por histiocitos epitelioides y células gigantes multinucleadas. Estos granulomas son más irregulares que los periglomerulares (más redondeados).

Figura 6. El caso de esta microfotografía corresponde al de un hombre de 62 años con poliangeitis microscópica. En una arteria intrarrenal evidenciamos esta lesión vascular. La pared arterial (flechas verdes) con su luz conservada (asteriscos) se ve claramente a la derecha; en la zona central se ve un área con necrosis fibrinoide de la pared, polimorfos y detritus celulares (flechas azules). Estas lesiones no son muy frecuentes en arterias intrarrenales, pero su presencia ayuda mucho en el diagnóstico de vasculitis. (Tricrómico de Gomori, X400).
En los túbulos pueden identificarse eritrocitos o cilindros eritrocitarios, pero su ausencia no implica que el paciente no ha tenido hematuria. Hay cambios de daño epitelial tubular en hasta el 57% de biopsias: descamación celular, pérdida del borde en cepillo, vacuolización citoplasmática y dilatación tubular; estos cambios se evidencian principalmente en pacientes con falla renal aguda. También pueden evidenciarse otros signos de daño crónico: atrofia tubular y fibrosis intersticial. En un 5% de casos hay franca inflamación tubulointersticial aguda. Hay fibrosis intimal de arterias, hipertrofia medial y depósitos arteriolares hialinos en una proporción variable de casos.
En pulmón hay extensa hemorragia alveolar, capilaritis y variable inflamación intersticial, tanto en enfermedad anti-MBG como en vasculitis sistémicas. El compromiso de otros órganos se caracteriza por la vasculitis de pequeños vasos, con la diferencia de encontrar granulomas en Wegener y marcado infiltrado de eosinófilos en Churg-Strauss.
Clasificación histopatológica de la GN asociada a ANCAs: Berden AE, et al. Histopathologic classification of ANCA-associated glomerulonephritis. J Am Soc Nephrol. 2010;21(10):1628-36. [PubMed link] [Free full text]
Inmunofluorescencia
En enfermedad anti-MBG hay depósitos lineales, globales y difusos de IgG (Figura 7), usualmente acompañados de menores cantidades de C3 lineal. Ocasionalmente hay tinción lineal para IgM o IgA en menor intensidad. Más raro aun es la presencia de anticuerpos de tipo IgA sin IgG. La IgG predominante es de subclase IgG1. Hay tinción lineal de IgG en basales tubulares en más del 50% de casos. También hay tinción lineal en la membrana basal de capilares alveolares, pero la tinción es más irregular, de menor intensidad y de más difícil interpretación dada su irregularidad, mayores artefactos y más tinción de fondo por plasma extravasado.
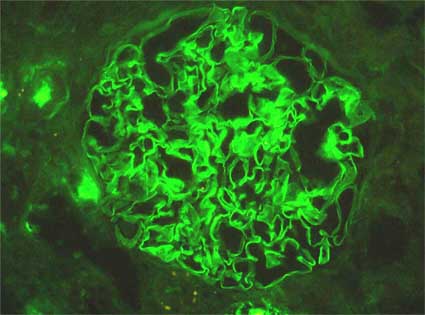
Figura 7. Inmunofluorescencia para IgG en un caso de síndrome de Goodpasture en un hombre de 29 años. Observe la tinción lineal en las paredes capilares. La inmunotinción para C3 puede mostrar un aspecto similar, pero suele ser menos intensa. (Inmunofluorescencia directa para IgG, anticuerpos anti-IgG humana marcados con fluoresceina, X400).
En la GNE pauciinmune (en cualquiera de los tres tipos de vasculitis o en la GNE idiopática) no se encuentran depósitos de inmunoglobulinas ni fracciones del complemento. En algunos casos pueden identificarse algunos depósitos débiles, pero si son intensos debemos pensar en enfermedad anti-MBG (lineales), o en enfermedad por complejos inmunes (granulares). En los segmentos necrotizantes puede haber atrapamiento de IgM y C3.
Las semilunas muestran positividad para fibrinógeno.
Microscopía electrónica
En enfermedad anti-MBG y GNE pauciinmune los hallazgos son muy similares: rupturas de la MBG y cápsula de Bowman, borramiento focal de procesos podocitarios, fibrina en espacio urinario y en el penacho y necrosis fibrinoide.
No debemos encontrar depósitos electrón-densos indicativos de enfermedad por complejos inmunes. En la enfermedad anti-MBG no hay este tipo de depósitos, sólo se evidencia una zona electrolúcida en el lado endotelial de la MBG correspondiente a edema.
Indicadores pronósticos
No hay hallazgos histopatológicos que específicamente indiquen mejor o peor pronóstico, pero, como en todas las glomerulopatías, las lesiones crónicas se correlacionan con daño irreversible y su extensión deberia cuantificarse o semicuantificarse: porcentaje de glomeruloesclerosis, de semilunas fibroepiteliales y fibrosas, grado de fibrosis intersticial, atrofia tubular y fibrosis arterial intimal. Además la severidad del daño glomerular: porcentaje de glomérulos con semilunas y necrosis, debe también determinarse. En último término es la evolución de cada caso individual la que determina el pronóstico. La clasificación de Berden AE et al. es útil para el pronóstico (Berden AE, et al.Histopathologic classification of ANCA-associated glomerulonephritis. J Am Soc Nephrol. 2010;21(10):1628-36. [PubMed link] [Free full text]).
Lecturas sugeridas: