
Homepage - - - Tutorial Index
Minimal change disease, IgM nephropathy, C1q nephropathy, IgG nephropathy and C3 mesangial nephropathy
Nephrotic syndrome (NS) associated to minimal glomerular changes:
“minimal change disease” (MCD) is a disease of unknown cause, that
predominantly affects children, and which there are not histologic alterations
with light microscopy or these are subtle. The diagnosis is confirmed by the
demonstration of podocyte foot process “effacement” or “fusion”.
Immunofluorescence is typically negative.
The disease has received several names: minimal change nephropathy, lipoid nephrosis, nil disease, and podocyte foot processes disease. The term “lipoid nephrosis” was used before the generalized use of the renal biopsy and suggested an alteration in the metabolism of lipids; the reason to use it was that in these patients oval fat bodies are seen in the urine and lipid droplets in some tubular cells in the autopsy.
There are primary (idiopathic) and secondary forms of the disease. The secondary forms can be the result of treatment with non-steroid antiinflammatory drugs, lymphoproliferative diseases (more frequently Hodgkin’s disease) and allergic reactions (like to bee bit).
It is difficult to determine the exact incidence of the disease because in children with NS biopsy is usually not carried out unless they do not respond to steroids or if the NS is corticodependent. It is calculated that 60% to 70% of children with NS will have MCD. It is, therefore, the most frequent cause of NS in this age group. MCD is more frequent in boys than in girls: 2:1, and the mean age of presentation is between 3 and 4-years-old; 80% of the children are under 6-years-old. In adults MCD corresponds to 10-20% of patients with NS and in this age group there is no the clear masculine predominance.
In opposition to focal and segmental glomerulosclerosis (FSGS), MCD occur more frequently in Caucasians than in Afro-American.
The prognosis is relatively good, usually respond to treatment with corticosteroids with disappearance of the ultrastructural alterations. In adults with NS a biopsy must be performed; in children the biopsy is usually carried out only if they are corticorresistant, corticodependent, or there are findings that suggest a different disease.
The etiology of MCD is not known. Although there are not immune deposits in glomeruli of these patients, an immune mechanism has been suspected to cause the disease. In many children the alteration is associated to a history of atopy, alimentary allergies and hypersensitivity reactions. Kark et al, in 1958, referred to MCD as “asthma of the nephron”. Numerous alterations of the humoral and cellular immunity have been described in patients with this disease. There is very little evidence of a humoral respose causing the disease, however, recently the cellular response has received more attention. Resolution of proteinuria with steroids and cyclophosphamide treatment in most of these patients suggest that cellular immunity has an important roll in the pathogenesis. Probably lymphokines produced by some T cells could generate, directly or indirectly, the ultrastructural and functional characteristic changes.
It is not completely clear how the structural alteration in podocytes causes proteinuria. In some works has been reported reduction in the ionic charge of the capillary wall, which could facilitate protein transport through the glomerular filtration barrier.
A great advance in the understanding of the idiopathic NS, and therefore of MCD and FSGS, was the discovery, in 1998, of the nephrin (NPHS1), the gene mutated in the congenital nephrotic syndrome of the Finnish type; this discovery was crucial to establish that the podocyte is the central component of the filtration barrier. Shortly after the gene mutated in corticosteroid resistant congenital nephrotic syndrome (NPHS2) led to the discovery of podocin, which is located solely in the slit-diaphragm region. In vivo we know that the nephrin is located in the slit diaphragm and interacts with podocin and with CD2-associated protein (CD2AP). This last one was known like a molecule related to the cytoskeleton of T cells. Nephrin and podocin are united to the cytoskeleton of the podocyte (transmembrane proteins) and their disruption not only affects the slit-diaphragm but also the integrity of the podocyte processes. Although not entirely clear yet, the physiopathogenesis of NS in MCD and FSGS affect this complex, and this change may be due to genetic defects (many have been described) or due to injury by immune or non-immune mediators (these are unknown). In the FSGS chapter we will discuss more about the proteins associated to the slit-diaphragm and the podocyte cytoskeleton, key structures to understand these diseases and NS in general. This structure and proteins are receiving very much attention and they are the center of many of the actual investigation in NS. (A good recent revisón in: Herve C, Dantal J. Possible new perspectives for our understanding of nephrotic syndrome recurrence. Nephrol Dial Transplant 21:10-13, 2006 [PubMed link / Free full text]. Another revision in: Tryggvason K, et al. Hereditary proteinuria syndromes and mechanisms of proteinuria. N Engl J Med. 2006 Mar 30;354(13):1387-401. [PubMed link] [Link to article in NEJM]).
Clinic features: The main manifestation is proteinuria, usually in nephrotic range producing edema. In many cases there are the other manifestations of the complete NS, but they can be absent. Hematuria is unusual and when present it usually is microscopic. Proteinuria is selective, unlike proteinuaria in FSGS and membranous GN (selectivity of proteinuria is determined by urine protein electrophoresis, although this test is not very often carried out in our center). There are some cases of MCD with very slight proteinuria and microscopic hematuria as the only findings, nevertheless is not clear if it is a “frustrated form” of the disease or, as some authors propose, “unspecific glomerular changes”. Usually there is not alteration in the renal function.
The clinical manifestations in adults are similar to those in children, nevertheless, there are a higher incidence of hypertension, nonselective proteinuria and renal failure.
Response to steroids is high in children: more than 90%. Many of these patients will present relapses that, in general, also respond to the treatment. The relapses tend to disappear before arriving at the adult age. The percentage of patients with complete remission, relapses, dependency of steroids and no-response to the treatment are very variable in the literature. Around 5% of children continue presenting relapses in the adult age, and less than 3% will develop renal failure at 10 post-diagnosis years. In corticoresistant patients, often, cytotoxic drug, as cyclophosphamide, are used with variable results.
In adults the steroid response is less than in children: 60-90% of cases. In addition, usually there is a greater time interval between the beginning of the treatment and the clinical response.
In secondary forms the prognosis depends on the underlying condition. The disease tends to disappear when disappear the associate cause.
Suggested adverse factors in the prognosis include: frequency of relapses, non-selective proteinuria, alteration of the renal function, and mesangial hypercellularity.
Laboratory features: Selective Proteinuria: albumin and other proteins of low molecular weight. The presence of high molecular weight proteins suggests another diagnosis. There is microscopic hematuria in 10-30% of patients. Usually there are normal values of BUN and serum creatinine. Complement leves are normal, and other tests that indicate formation of immune complexes are normal.
Histopathology
By definition, there are not glomerular histologic changes or these are subtle (slight mesangial proliferation). As in any cause of NS, droplets of protein resorption may be seen in the cytoplasm of podocytes or tubular cells, and in some cases podocyte hypertrophy may be evidenced. Cellularity is normal in most of cases (Figure 1 and Figure 2). In some cases there is slight mesangial hypercellularity, and it is discussed if this finding is correlated with a less steroid responsive, some works have not found such relation. When there is hypercellularity is more probable to find IgM deposits (IgM nephropathy, see below) (Figure 3 and Figure 5). There are nor glomerular segmental lesions, if present we must not diagnose MCD.
Figure 1. Renal biopsy of a 16-years-old man with nephrotic syndrome and MCD diagnosis. See tuft cellularity and the normal aspect of the glomerulus. Immunofluorescence was negative. (H&E, X400).
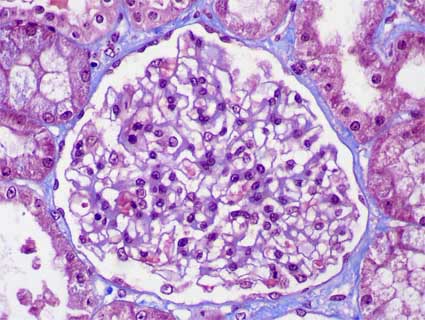
Figure 2. Another case of MCD in which we see the normal appearance of the glomerular tuft, there is not hypercellularity (Masson’s trichrome, X400).

Figure 3. Renal biopsy of a 10-years-old girl with NS; the immunofluorescence was negative and the ultrastructural study showed foot processes effacement (“simplification”), without electron-dense deposits. See the mesangial hypercellularity, more accentuated in the lobes pointed with the arrows. This case was diagnosed as MCD with mesangial hypercellularity. The NS was initially resistant to steroids, but a year later proteinuria had diminished to non-nephrotic range. (H&E, X400).
Some glomeruli can be globally sclerosed, like in any normal kidney; the traditionally used formula to determine the maximal “permitted” percentage of sclerosed glomeruli is: age ÷ 2 - 10; this formula is applied to adults; in children we can see only occasional sclerosed glomeruli. Any segmental lesion (it can be very subtle) must to raise the possibility of another diagnosis, as FSGS (Figure 4).
In children there are not tubulointerstitial alterations. If there is fibrosis and/or tubular atrophy another disease must be suspected (usually FSGS). In normal adults is frequent to find some small areas of interstitial fibrosis and tubular atrophy, therefore in them this alteration is not incompatible with MCD.
Just as in the tubulointerstitium, in vessel there are not alterations.

Figure 4. In this case of a 5-years-old boy with corticorresistant NS we did not find tubulointerstitial lesions and the immunofluorescence was negative; almost all the glomeruli had a histologic normal aspect, but in one (in the microphotography) we found a sinequia of the tuft to the Bowman’s capsule. These alterations, even being subtle, indicate some degree of glomerular injury that must make us raise the possibility of other glomerulopathy, as FSGS. (Methenamine-silver, X400).
See case 71 of our Case Series: Hipercellular "variant" of minimal change disease.
Immunofluorescence
There are neither immunoglobulins nor complement fractions deposition in renal tissue. In some cases there are weak deposits of C3 in mesangium; this feature does not correlate with clinical presentation or outcome. When there is intense mesangial deposits of IgM or C1q probably there are pathogenic implications and is possible that clinical outcome to be diverse to MCD (see below).
Electron microscopy
The diagnostic ultrastructural changes are between the podocyte and the glomerular basement membrane. Architecture of the podocyte processes is lost, slit-diaphragms are not identified. This finding is called “effacement”, “fusion”, or “simplification” of podocyte processes, although these are misnames; this change results from disordered epithelial cell structure with withdrawal of the dendritic process (Figures 4b, 4c, 4d). The cytoplasm of podocytes displays long and narrow prolongations from its surface: “microvillous transformation” (Figure 4c). It is common for the cytoplasm to appear more electron-dense in the part adjacent to the glomerular basement membrane: cytoskeletal condensation (Figure 4b). Podocyte alteration is usually diffuse, nevertheless, there are cases which this finding is focal. There is not a constant correlation between the degree of ultrastructural injury and the severity of proteinuria. These changes revert when there is response to the treatment. (Several electron microscopy images of MCD in AJKD (link)])
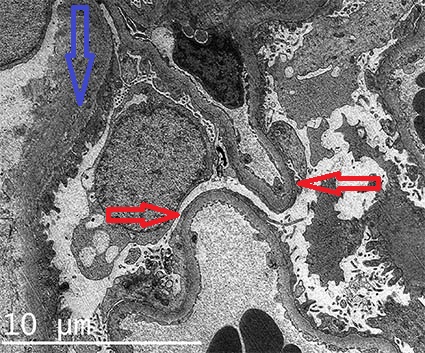
Figure 4b. Diffuse podocyte damage, note loss of pedicels and slit diaphragm (red arrows). The basement membranes have normal features. The blue arrow points to the basal membrane of Bowman's capsule (Electron microscopy, original magnification, X1,500.

Figure 4c. Podocyte damage with complete loss of podocyte processes- Note the cytoplasm of the podocyte a little more dense in the portion closest to the basement membrane: cytoskeletal condensation. The basement membrane has normal features (Electron microscopy, original magnification, X4,000).

Figure 4d. In this image, in addition to podocyte damage, thin and elongated processes of the podocyte cytoplasm are observed: microvillous transformation of the podocyte cytoplasm (arrows) (Electron microscopy, original magnification, X2,500).
See Case 174 of our Case Series: Minimal change disease in the elderly.
When there is intense and diffuse mesangial deposits of IgM, associated to NS, many authors consider that it is a disease different to MCD and FSGS; nevertheless, others consider that it is a variant of MCD or FSGS.
Immunoglobulin M nephropathy (IgMN), known since 1978, is a very controversial clinicopathological entity characterized by IgM diffuse deposits in the mesangium at immunofluorescence whereas light microscop identifies minimal glomerular lesion, hypercellularity and expansion of the mesangium or sclerotic focal, segmental lesion. Clinically, it is a nephrotic syndrome, especially in pediatric patients, or asymptomatic proteinuria and/or isolated hematuria. These characteristics narrowly define IgMN between minimal change disease and focal segmental glomerulosclerosis, so it is not often recognized as a separate pathology. Homogeneous epidemiologic, pathogenetic, clinical or histological data are not available (Brugnano R, et al. IgM nephropathy: is it closer to minimal change disease or to focal segmental glomerulosclerosis? J Nephrol. 2016 Aug;29(4):479-86. [PubMed link]; Arias LF, et al. IgM nephropathy in children: clinicopathologic analysis. Nefrologia. 2013;33(4):532-8 [PubMed link]).
The clinical presentation is very similar to that of MCD, but it seems that there is minor response to steroids and the prognosis can be worse (Myllymaki J, et al, IgM nephropathy: clinical picture and long-term prognosis. Am J Kidney Dis;41:343-50, 2003 [PubMed link]).
Many cases display mesangial hypercellularity (Figure 5).
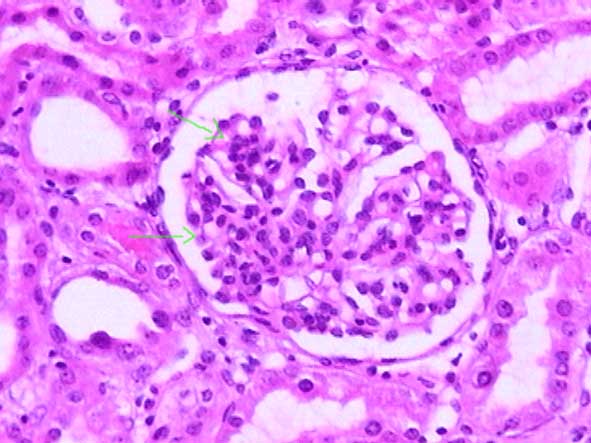
Figure 5. Renal biopsy of a 5-years-old boy with corticodependent NS. The glomeruli showed mesangial hypercellularity; there were not tubulointerstitial or vascular alterations. By immunofluorescence we demonstrated diffuse and global mesangial IgM deposits, with C3 in less intensity and similar location. There were not glomerular segmental lesions. It was diagnosed as IgM nephropathy (H&E, X300).
See Case 57 of our Case Series for a typical case with a more extensive discussion.
It is a glomerulopathy in which there is intense and diffuse mesangial C1q deposition, accompanied generally by mesangial deposits of other immunoglobulins and complement fractions. Histologic features vary from no changes (as in MCD) to mesangial cellular proliferation or focal and segmental sclerosing lesions.
The usual clinical presentation is severe proteinuria, the majority with the other features of NS, proteinuria does not revert with steroids or the patients present multiple relapses. In many cases there is hematuria. Most of patients are young or adult young and there is predominance of Afro-American. Many cases seem to be within the clinical spectrum and evolution of MCD/FSGS (Tariq N, et al. C1Q Nephropathy: A Multifaceted Disease With Infrequent Diagnosis. J Ayub Med Coll Abbottabad. 2019 Jul-Sep;31(3):308-313. [PubMed link]; Kim K, et al. C1q nephropathy in adults is a form of focal segmental glomerulosclerosis in terms of clinical characteristics. PLoS One. 2019;14(4):e0215217. [PubMed link]; Devasahayam J, et al. C1q Nephropathy: The Unique Underrecognized Pathological Entity. Anal Cell Pathol (Amst). 2015;2015:490413 [PubMed link]; Markowitz GS, et al, C1q nephropathy: a variant of focal segmental glomerulosclerosis. Kidney Int 64:1232-40, 2003. [PubMed link]; Kersnik Levart T, et al, C1Q nephropathy in children. Pediatr Nephrol 20:1756-61, 2005. [PubMed link]).
There are not systemic autoimmune disease nor hypocomplementemia. Renal function is not affected in initial stages of the disease.
It is not clear the prognosis, but some patients evolve to terminal renal failure. [Several images and EM (AJKD - link)]
Although more studies are needed to determine the physiopathogenic mechanism of the C1q deposits, it is possible that these arise by an unspecific deposition when increasing the passage of these molecules to mesangium in the context of proteinuria (Markowitz GS, et al, C1q nephropathy: a variant of focal segmental glomerulosclerosis. Kidney Int. 64:1232-40, 2003. [PubMed link]).
Seeer Case
10 of our case series.
A very good recent article: Fukuma Y, Hisano S, Segawa
Y, Niimi K, Tsuru N, Kaku Y, Hatae K, Kiyoshi Y, Mitsudome A, Iwasaki H. Clinicopathologic
Correlation of C1q Nephropathy in Children. Am J Kidney Dis. 2006;
47:412-8 [PubMed
link]
This is a entity of relatively recent description, not described in many texts and, possibly, not accepted by all authors, since it is not very well defined and characterized.
IgG nephropathy (or mesangial IgG glomerulonephritis) has seen described in patients with NS and patients with hematuria and proteinuria, without NS. In most of cases there are also deposits of C3. There is doubt if it is a different disease or it is a variant of MCD. Due to the small number of reported cases its prognosis is not clear, but apparently, al least half of the patients evolve to chronic renal failure. (Fakhouri F, et al, Mesangial IgG glomerulonephritis: a distinct type of primary glomerulonephritis. J Am Soc Nephrol. 13:379-87, 2002. [PubMed Link / Free full text])
Some authors have reported that there is an overlap between IgG nephropathy and C1q nephropathy, for which they propose the term "IgG / C1q nephropathy" (Lim BJ, Hong SW, Jeong HJ. IgG nephropathy - confusion and overlap with C1q nephropathy. Clin Nephrol. 2009 Nov;72(5):360-5. [PubMed Link]). We will have to wait for more studies and case series to have more clarity about these alterations.).
Bibliography
Homepage - - - Tutorial Index