
Homepage - - - Tutorial Index
IgA Nephropathy and Schönlein-Henoch
Purpura
Pathogenesis - Clinical features - Histopathology - Henoch-Schönlein purpura
IgA nephropathy (IgAN) is defined by the presence of diffuse dominant or codominant mesangial deposits of immunoglobulin A (IgA). The histologic aspect is very variable, being more frequent mesangial alterations: cellular and/or matrix proliferation. Clinically the most common findings are microscopic hematuria (persistent or intermittent) and episodic macrohematuria. Although in the medical literature before the Sixties there are descriptions of alterations that could correspond to IgAN, is only with the works of Berger and Hinglais in 1968 and 1969 that the disease was well recognized and classified. IgAN is also known as Berger’s disease, IgA mesangial glomerulonephritis, IgA mesangial nephropathy.
IgAN may be primary or secondary to other diseases, mainly hepatobiliary disease of any cause. Other diseases associated with IgAN are: celiac disease, Crohn’s disease, ulcerative colitis, Sjögren’s syndrome and other rheumatologic diseases, chronic infectious or inflammatory mucosal diseases (specially of lung), autoimmune diseases as dermatitis herpetiformis, lymphoproliferative diseases, carcinoma of colon, stomach, breast, and lung.
Schönlein-Henoch (S-H) purpura, also known as S-H syndrome, S-H nephritis, anaphylactoid purpura, and rheumatoid purpura (in some texts the eponymous appears inverted: Henoch-Schönlein), is characterized by purpuric skin lesions (mainly on the lower extremities and buttocks), migratory arthralgias, gastrointestinal hemorrhage and renal affectation. The syndrome, described by Johann Lukas Schönlein (1793-1864) in 1837 and Eduard Heinrich Henoch (1820-1910) in 1867, is considered a form of IgAN with prominent extrarrenal involvement: systemic vasculitis mediated by IgA rich immune complexes (see below).
IgAN is recognized like the more frequent primary glomerulonephritis in the world and it is cause of terminal renal failure in approximately 10-15% of all the patients who arrive at this state. The disease is commonest in the south of Europe, Asia and in native Americans. It is diagnosed in 5-44% of all renal biopsies according to different series: 5-10% in North America and United Kingdom, 20-35% in some European series, and 25-45% in Asia. The policies or protocols with respect to taking renal biopsies respond on part for this variability; in centers where cases of isolated microhematuria usually are not biopsied the incidence will be smaller; and in regions where control with urinalysis is performed regularly in asymptomatics there will be greater percentage of cases. Some works suggest that prevalence of subclinical IgAN is much more high of which it seems.
IgAN occurs in all ages but is most frequent (approximately 65% of patients) in the second or third decades of the life; the mean age of diagnosis is 28-years-old; less than 15% of patients are over 40-years-old, and it is not very common in <10-years-old. The ratio male:female is approximately 2:1, but in series from Asia the relation is near 1:1, and in North America and Europe it is of 3,1:1. IgAN affects all the races, and there is a higher incidence in native Americans. In some series it seems to have less incidence in African and Afro-American, nevertheless, this data could be due to smaller access to medical attention and greater incidence of other more severe renal diseases (Sehic AM et al, Pediatr Nephrol 11:435-7, 1997 [PubMed link]).
The development of IgAN appears to require an autoantibody (IgG or IgA) against the autoantigen galactose-deficient IgA1 (Wyatt RJ, Julian BA. IgA nephropathy. N Engl J Med. 2013;368(25):2402-14). Pathologically, IgAN is an IgA dominant or codominant immune complex–mediated glomerulonephritis that may have codeposition of IgG by immunofluorescence microscopy (IF). However, like in many other glomerulonephritis, the cause of IgAN is not clear, although we know some pathogenic aspects. Mesangial deposits are predominantly of polymeric IgA , subclass 1 (pIgA1).Current evidence suggests that IgA nephropathy is not due to a single pathogenic insult, but rather the result of multiple sequential pathogenic "hits". An abnormally increased level of circulating poorly O-galactosylated IgA1 and the production of O-glycan-specific antibodies leads to the formation of IgA1-containing immune complexes, and their subsequent mesangial deposition results in inflammation and glomerular injury. While this general framework has formed the foundation of our current understanding of the pathogenesis of IgA nephropathy, much work is ongoing to try to precisely define the genetic, epigenetic, immunological, and molecular basis of IgA nephropathy. In particular, the precise origin of poorly O-galactosylated IgA1 and the inciting factors for the production of O-glycan-specific antibodies continue to be intensely evaluated. The mechanisms responsible for mesangial IgA1 deposition and subsequent renal injury also remain incompletely understood (Yeo SC, et al. New insights into the pathogenesis of IgA nephropathy. Pediatr Nephrol. 2018;33(5):763-777).
Three key elements are related to development, severity and evolution of IgAN (Barratt J y Feehally J, J Am Soc Nephrol 16:2088-97, 2005 PubMed link): 1.) Synthesis, liberation and persistence in the circulation of pIgA1 with characteristics that favor their mesangial deposit; 2.) The “reactivity” of mesangium: more o less susceptibility to pIgA1 deposition and more o less capacity to generate an inflammatory response to the deposits; and 3.) the response of the kidney to the aggression and its tendency to initiate and perpetuate a cascade of events that favors greater injury (tubule-interstitial damage and glomerulosclerosis) and not the resolution of the process. Each one of these three elements can have a genetic base that influences the phenotype of the disease in each patient.
Many alterations in the IgA have been described and in their production, nevertheless, the alterations are heterogeneous in different groups of patients. This concept supports the hypothesis that there are several pathogenic mechanisms of the disease. Only the increase of the circulating IgA is not sufficient to produce mesangial deposits; other special characteristics are necessary to promote its location there.
It is not demonstrated that IgA is an autoantibody against mesangial antigens. On the contrary, many circulating IgA1 antibodies against a variety of antigens have been described, but no single pathogenic antigen has been identified. Studies of serum IgA have, by contrast, demonstrated a number of unusual physical characteristics in IgAN, but it is not known which of these is responsible for the deposition in mesangium and for activation of mesangial cells. One of the more studied structural alterations is the abnormal glycosylation of IgA1 molecules. Current in vitro evidence suggests that aberrantly glycosylated IgA1 molecules have an increased tendency both to self-aggregate and to form antigen-antibody complexes with IgG antibodies directed against IgA1 hinge epitopes, favoring the generation of macromolecular aggregates of pIgA1 and IgA immune complexes (IgA-IC), which would promote mesangial deposition. In addition, this type of altered IgA1 molecules has more affinity, in vitro, by components of the extracellular matrix: fibronectin and type IV collagen.
The site of production of pathogenic IgA in IgAN has been object of multiple studies. The association between episodes of macrohematuria and upper respiratory tract infections or other mucosal inflammations allow suspect the presence of abnormal mucosal antigens, particularly because both mesangial IgA and the increased IgA fraction in serum IgA are polymeric, which is normally produced at mucosal surfaces rather than in systemic immune sites. However, studies indicated that mucosal pIgA plasma cell numbers are normal or even reduced in IgAN, whereas pIgA antibody levels in mucosal secretions are not elevated and are sometimes lower than controls. However, increased pIgA1 plasma cell numbers are found in the bone marrow in IgAN, and systemic antigen challenge results in increased titers of circulating pIgA1 antibodies, with normal levels in mucosal secretions. Therefore, the overproduction of pIgA1 is likely to be based within systemic immune sites such as the bone marrow, with both systemic and mucosal antigen challenges resulting in aberrant systemic immune responses (Barratt J y Feehally J, J Am Soc Nephrol 16:2088-97, 2005 [PubMed link]). Therefore the overproduction would be a systemic event, probably originated in response to mucosal antigens. Reinforcing this concept, we know that the IgA produced in mucosal is predominantly IgA2 and the one that is deposited in glomeruli is IgA1.
Also an alteration in the elimination of IgA and IgA-immune complexes has been proposed, which would facilitate its persistence in the circulation. One of the mechanisms of degradation of IgA is by a Fc receptor: CD89, which mediates their endocytosis and catabolism. In IgAN there is a subregulation of CD89 in myeloid cells what would reduce its elimination. Also a less affinity of IgA by the CD89 receptor has been demonstrated in some cases of IgAN.
All the IgA deposits do not generate inflammation nor these mesangial deposits are irreversible. It is possible that the increase of IgA macro-molecules facilitates its unspecific trapping in mesangium, perhaps it is increased by the glycosylation defect of the molecule. Also, the formation of free CD89-IgA complexes in the circulation that would be deposited in mesangium has been suggested.
There is in vitro evidence that human mesangial cells are capable of receptor-mediated endocytosis and catabolism of IgA. It is possible that some alteration in the degradation or clearance mechanism contribute to IgA accumulation and IgAN development.
IgAN is not, in general, associate with inflammatory migration of cells to mesangium, suggesting that glomerular injury is due mainly to resident mesangial cells response. The glomerular injury happens predominantly through activation of the complement induced by IgA. IgA mesangial deposition overregulates the synthesis of extracellular matrix and an proinflammatory and profibrotic state of mesangiales cells. Although complement activation is not essential for IgAN development, there is evidence that suggests its activation can influence the extension of the glomerular damage. C3 activation by mesangial IgA allow generation of C5b-C9 that would activate the mesangial cell generating production of mediators of the inflammation and components of the extracellular matrix. Like in lupus nephritis, C3 deposited in mesangium is in activated form, emphasizing the importance of the activation of the complement in this disease.
There are genetic factors related to the development of the disease and the existence of familial forms is well-known. Nevertheless, until now there are no genes candidates. Studies in relation to different genes and loci will give some light us, hopefully near in the future.
IgN commonly recurs in allografts, although unusually it is clinically significant; and in kidneys from donors with IgA mesangial deposits these disappear post-transplant; these data indicate some underlying extrarrenal factor in the nephropathy.
Secondary IgAN: in cases of IgAN secondary to hepatic disease, the roll of liver in clearance of IgA molecules, mainly IgA1, seems important, the hepatocytes are the main route of IgA degradation. In addition to serum IgA increase, other mechanisms play a important role in the pathogenesis of IgN secondary to hepatobiliary disease: alteration in monocyte phagocytosis, accumulation of other nocive substances, greater risk of infection, hypocomplementemia and increase in TGF-beta production.
In IgAN associated to mucosal diseases, overproduction of IgA seems to play a great role, nevertheless, other mechanisms, few understood, must be important in the pathogenesis; perhaps production of some cytokines or alterations of the mucosal barrier to the passage of antigens. In IgAN secondary to disorders of the immunity and lymphoproliferative diseases the pathogeny is not clear; disorder in the production of growth factors, alterations of T-cells, altered production of cytokines, and other mechanisms, have been implicated in the pathogenesis.
Clinical features: Hematuria is the clinical manifestation more consistently expressed in IgAN, it is observed at some time of the evolution in more than 95% of patients. Persistent microhematuria is found in approximately 78% of cases and episodic macrohematuria in 54%. Macrohematuria usually appears by few days and microhematuria between these episodes is detected. In other cases may be intermittent microhematuria. In some series, macrohematuria is more frequent in children, while other series find little difference between adults and children. Another frequent feature is proteinuria, usually <1g/24h, although 5-10% of patients will have severe proteinuria, or they will even develop complete nephrotic syndrome. There is hypertension in ~15% of patients, and serum creatinine increase in ~10%. In ~5% of cases chronic renal failure at diagnosis is detected. It is less probable to find HTA and impairment of renal function in children. In few patients there is abdominal or flank pain.
In many cases in which IgAN appears like episodic macrohematuria, the episode appear associate to pharyngitis; unlike the postinfectious GN, in IgAN hematuria occurs shortly after pharyngitis: sinpharyngitic; separation between pharyngitis beginning and hematuria is days, more than weeks like in postinfectious GN (postpharyngitic). Episodes of gastroenteritis, sinusitis or bronchitis are also associate with hematuria. In spite of having, at least in some cases, more severe histologic glomerular changes in episodes of macrohematuria, some works show better prognosis for patients with this clinical manifestation.
In some patients there is associate acute renal failure to episodes of hematuria; this renal failure usually is transitory and it does not seem to affect the prognosis; in many cases it is associate to tubular damage due to hemoglobin. In other cases it may be associated to extracapillary proliferation (crescents) and rapidly progressive GN, with a worse prognosis: greater probability of evolution to chronic renal damage. In the patients with persistent or episodic microhematuria there is less incidence of acute renal failure, but greater probability of progressive diminution of the glomerular filtration rate.
In patients with IgAN and nephrotic syndrome a collision between IgAN and minimal change disease (MCD) has been proposed, nevertheless, although usually it is impossible in this context to verify this coexistence of diseases, the number of such patients exceeds the spected according to the frequency of MCD. Easier to verify has been the collision of membranous GN and IgAN, although it is an unusual event. Nephrotic syndrome in IgAN could be the result of a physiopathogenic different process.
Although initially considered a benign disease, we know that IgAN may evolve to terminal renal failure in approximately 30% of cases to 20 years of the diagnosis. In many cases there is spontaneous remission, but it is maintained in only ~10% of cases. Between 60% and 70% of cases will continue with episodes of worsening and relative calm of the disease. IgAN is the cause of terminal renal failure around 10-15% of all the cases.
IgAN recurs post-transplantation in approximately 50% of cases: as deposits of IgA, nevertheless, the risk of significant dysfunction is of ~13% and the one of renal failure ~5% to five years.
There is no a well established effective therapy for the disease. The control of underlying conditions like HTA is important. Steroids have few utility, although in patients in whom there is nephrotic syndrome a greater proportion of response is reported. Calcium channels blocking agents and angiotensin-converting enzyme inhibitors seem be useful in patients without HTA. Although rich Omega-3 fish oil have gained acceptance, there are still no many studies to support its role, and other works did not suggest efficacy. Immunosuppressors and other treatments have been used with variable results. In some cases tonsillectomy is recommended. In many patients any treatment is not provided, and this patients are periodically controlled.
Laboratory findings: There is hematuria, at some time of the evolution, in 95% of cases. Proteinuria is very variable, but it is usually <1g/24h, serum creatinine and BUN are in normal levels in most of the patients. Serum levels of IgA may be increased in many patients, but this increased level does not constitute a test with high sensitivity or specificity for the diagnosis. There are no alterations in complement levels.
The histopathologic features are very variable, from normal appearing glomeruli to any type of proliferative or sclerosing lesion, including extracapillary proliferation. The predominant glomerular changes are mesangial proliferation (cells and matrix) and focal proliferative nephritis; IgAN is the first possibility when we find this last histologic change in a patient without systemic disease. Mesangiopathic glomerulopathy is the morphologic presentation in more than 60% of cases; in general, there is increase of cells and mesangial matrix in variable degrees. The alteration tends to be more or less uniform in all the glomeruli, but in some cases there is great variation among them in a same biopsy. Mesangial expansion is more appreciated with PAS stain that with methenamine-silver stain due to more affinity of mesangial deposits for PAS reactive; the degree of this expansion is difficult to determine accurately, is something subjective and with little reproducibility among observers, unless it is measured by morphometric computer analysis. The immune deposits usually are not seen with light microscopy.
Figure 1. Glomerular tuft with mild global mesangial hypercellularity; direct immunofluorescence demonstrated diffuse mesangial IgA deposits. This case correspond to a 29-years-old woman with recurrent macrohematuria and heavy proteinuria, renal function did not present alterations (H&E, X400).
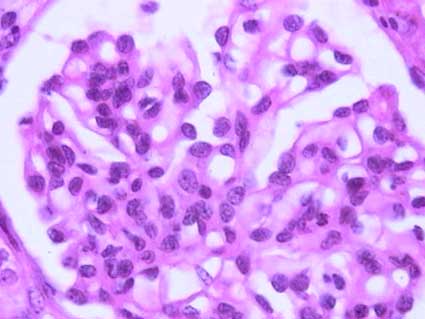
Figure 2. Mesangial hypercellularity is seen more clearly in this glomerulus of a 45-years-old patient with IgA nephropathy. (H&E, X400).
In some cases the mesangial lesions are focal and segmental. There is focal and segmental endocapillary proliferation, with infiltration of leukocytes, in approximately 17% of cases, and in almost 5% there is diffuse endocapillary proliferation. In a variable proportion of cases segmental and focal sclerosing lesions are identified; there can be tuft adhesions to Bowman’s capsule and segments with podocyte hypertrophy and hyperplasia. In few cases there are segmental necrosis in the glomerular tuft. We can see crescents in glomeruli with any other type of glomerular change; in 5-8% of patients crescents involve more than 50% of glomeruli. Frequently there are subepithelial deposits associated to the crescents. We can also see other patterns of glomerular change in IgAN: membranoproliferative, membranous, and mixed glomerulonephritic lesions. Around 12% of cases show normal or nearly normal glomeruli.
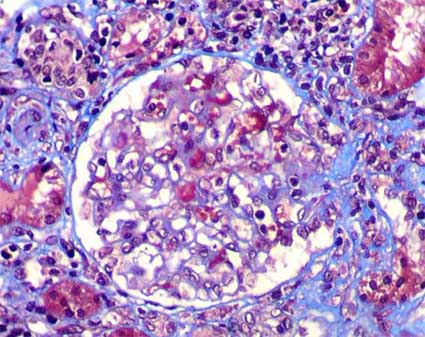
Figure 3. In some IgAN cases we will find segmental glomerular sclerosing lesions like in this biopsy of a 37-years-old man with persistent microhematuria. These changes may indicate a greater degree of severity of renal alterations, but, not necessarily they are correlated with proteinuria or renal function. (Masson’s trichrome, X400).
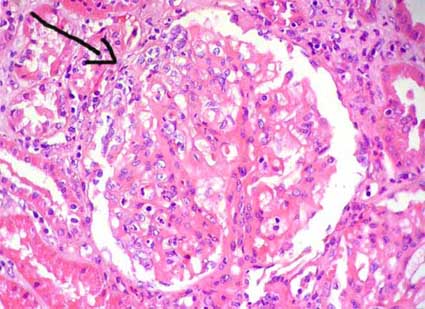
Figure 4. In this glomerulus, in addition to mesangial hypercellularity, there are segments with endocapillary proliferation, inflammatory cells, and diminution of capillary lumina. The arrow indicates a small epithelial circumscribed crescent. (H&E, X400).
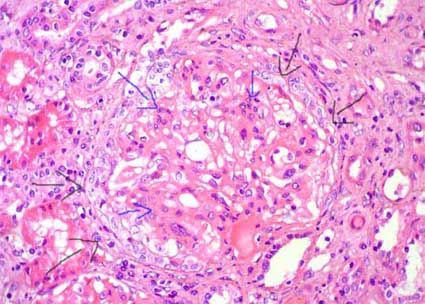
Figure 5. Same case of the previous microphotography; see the tuft compressed by a circumferential epithelial crescent (black arrows); there are cellular and matrix mesangial proliferation (blue arrows). This case corresponds to a 32-years-old woman with episodes of macrohematuria; in the last episode, when this biopsy was undertaken, she presented, in addition, serum creatinine increase. Extensive crescentic glomerulonephritis in IgAN is associated with more aggressive clinical outcome. (H&E, X400).
In tubules it is possible find erythrocytes or erythrocyte casts, but its absence does not imply that the patient has not hematuria. There are changes of tubular epithelial damage in until 57% of biopsies: cellular desquamation, loss of the microvillous brush border, cytoplasmic vacuolization, and tubular dilatation; these changes are demonstrated mainly in patients with acute renal failure, and may be due to damage by hemoglobinuria. Changes of chronic damage can be demonstrated: tubular atrophy and interstitial fibrosis. In 5% of cases there is frank acute tubulointerstitial inflammation. There are medial intimal fibrosis of arteries and hypertrophy and hyaline deposits in arterioles in a variable proportion of cases.
Several systems to classify the microscopic changes in IgAN has been developed, nevertheless, until the moment there is no an universally accepted classification; different works show variable correlation with the prognosis. Some of the most used are the one of S.R. Meadow et al (Meadow SR, et al, Q J Med 41:241-58, 1972[PubMed link]), the one of M. Haas (Haas M, Am J Kidney Dis 29:829-42, 1997[PubMed link]), the one of H.S. Lee et al (Lee HS, et al, Nephrol Dial Transplant 20:342-8, 2005 [PubMed link]) and the one of S.M.K. Lee et al (Lee SM, et al, Hum Pathol 13:414-22, 1982 [PubMed link]). These classifications are based on the severity and extension of mesangial proliferation, the percentage of glomeruli with segmental lesions, crescents, and global sclerosis, and, in some schemes, the extension of the tubulo-interstitial damage. The International IgA Nephropathy Network developed a proposal of a new classification of the IgAN with better interobserver reproducibility, and that defines better the types and degrees of injury, this work was supported by the Renal Pathology Society and the International Society of Nephrology and appears in two papares: "rationale, clinicopathological correlations, and classification" y "pathology definitions, correlations, and reproducibility".
Oxford Classification of IgA nephropathy
Initially published in 2009 and modified in 2016 (adding the "C" score) it graduates 5 parameters: mesangial hypercellularity (M), endocapillary hypercellularity (E), segmental sclerosing glomerular lesions (S), interstitial fibrosis and tubular atrophy (T) and crescents (C):
Oxford Classification of IgA nephropathy (MEST-C) (Cattran DC, et al. The Oxford classification of IgA nephropathy: rationale, clinicopathological correlations, and classification. Kidney Int. 2009;76:534-45 [PubMed link]; Trimarchi H, et al. IgAN Classification Working Group of the International IgA Nephropathy Network and the Renal Pathology Society; Conference Participants. Oxford Classification of IgA nephropathy 2016: an update from the IgA Nephropathy Classification Working Group. Kidney Int. 2017;91:1014-1021 [PubMed link]). |
|
M |
M0: There is no mesangial hypercellularity or it compromises <50% of glomeruli M1: more than half the glomeruli have more than three cells in a mesangial area |
E |
E0: Absent: No endocapillary hypercellularity E1: Hypercellularity due to increased number of cells within glomerular capillary lumina causing narrowing of the lumina |
S |
S0: Absent: There are no segmental sclerosing glomerular lesions S1: Any amount of the tuft involved in sclerosis, but not involving the whole tuft or the presence of an adhesion |
T |
T0: Interstitial fibrosis or tubular atrophy (IF/TA) in < /= 25% T1: IF/TA in 26-50% T2: IF/TA >50% |
C |
C0: There are no crescents C1: Crescents in <25% (cellular or fibrocellular*) C2: Crescents in >/=25% (cellular or fibrocellular*) |
*Fibrous crescents (composed of >/=90% matrix) are not taken into account.
As can be seen, more than a classification, it is a semi-quantitative classification of 5 parameters. From a personal point of view, it is a "classification" that leaves out a lot of important information, for example, it is not the same if in a biopsy we find 1 glomerulus out of 40 with segmental sclerosis, than another in which we find 20 out of 40 with said segmental sclerosis, in both cases the "S" parameter is S1, however, we all know that with so many compromised glomeruli the prognosis will be worse. This classification also does not take into account the percentage of global glomerulosclerosis, which, logically, is very important as a prognostic factor. It also does not take into account the presence of necrotizing lesions. More important than a grading or classification, to define the management of a patient it is essential to analyze the entire clinical context and the detailed information of a good report of the biopsy study (the same for all classifications in renal pathology).
Immunofluorescence
IgA mesangial deposits is the characteristic and defining feature of the disease, nevertheless, the deposits are exclusive of IgA in only ~26% of cases; usually they are accompanied by IgG (~37%) or IgM (~13%), and the three Igs. may be present in ~25% of cases. By definition, IgA must be dominant or codominant; if there are deposits of IgG or IgM more intense than those of IgA we must think in another glomerulopathy. There are C3 deposits in the great majority of cases (~95%) and occasionally components of the classic pathway of complement activation: C1q and C4 (~12%). IgA deposits are granular mesangial diffuse in almost all the patients, nevertheless, occasionally they are segmental. The predominant IgA subclass is 1 and in terms of light chains lambda predominates. There is not deposition of secretory component of IgA. In most of patients membrane attack complex (C5b-C9) is detected but its physiopathogenic importance is not very clear. Some studies have shown that the presence of C4d deposits have been related to lower renal survival (Espinosa M, et al. Association of C4d deposition with clinical outcomes in IgA nephropathy. Clin J Am Soc Nephrol. 2014;9(5):897-904 [PubMed Link], Segarra A, et al. Mesangial C4d Deposits in Early IgA Nephropathy. Clin J Am Soc Nephrol. 2018;13(2):258-264 [PubMed link].
In approximately 25% of cases there are variable degrees of IgA deposits in capillary walls. IgG co-deposition and the location of glomerular immune deposits in the capillary walls appear to be both associated with greater histologic activity on renal biopsy, but only the location of glomerular immune deposits in the capillary walls was associated with a significantly increased risk for end-stage renal disease, transplant, death and/or doubling of SCr in a study (Alvarado AS, et al. Location of glomerular immune deposits, not codeposition of immunoglobulin G, influences definitive renal outcomes in immunoglobulin A nephropathy. Nephrol Dial Transplant. 2018;33(7):1168-1175. [PubMed link]).
See Case 24 of our Case Series. IgA nephropathy with IgM co-deposits and subendothelial deposists
See Case 153 of our Case Series: Subendothelial depósitis and IgG co-deposition.

Figure 6. The fundamental characteristic in IgAN is the intense, diffuse mesangial immunostaining for IgA. See that positivity is limited, in this case, to mesangial areas, without deposits evidenced in capillary walls; this microphotography represents the typical mesangial pattern (Immunofluorescence with anti-IgA antibodies marked with fluorescein, X400).
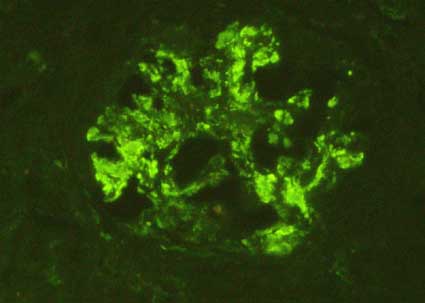
Figure 7. When mesangial widening is notorious, the positivity is more extensive within the glomeruli. Sometimes we can also see some parietal deposits (in capillary walls) (Immunofluorescence with anti-IgA antibodies marked with fluorescein, X400)..
Electron microscopy
There are electron-dense mesangial deposits that correlate with the immune deposits. Many of these are located in the interphase between mesangium and capillary lumina, or at the union of mesangium and GBM: paramesangium. These deposits have a variable size; some authors think that the deposits located deeper in mesangium reflect more advanced stages of the glomerulopathy. There are subendothelial deposits in approximately 19% of cases, subepithelial deposits in ~12% and intramembranous deposits in ~13% of the cases. Mesangial cells present increase of mitochondries and rough endoplasmic reticulum.
In some cases lamination and fragmentation of the GBM similar to the changes seen in Alport’s disease are detected, and other works have documented thin basement membranes disease in cases of IgAN. The coexistence of these changes has clinical and prognostic uncertain implications; their pathogenesis is not very clear; it could be a collision of two glomerular diseases.
Prognostic markers
Clinical characteristics associated with greater probability of chronic renal damage include: increasing age at presentation, no presence of macrohematuria episodes, severe proteinuria (in some studies >1g/24h), hypertension, persistent diminution of glomerular filtration rate, serum creatinine >1,4 mg/dL, and diminished urine concentration capacity. Recurrent macroscopic hematuria is a good prognosis indicator. No impact on prognosis is found for gender, ethnicity, and IgA serum levels.
Histological alterations related to a worse prognosis: extension of interstitial fibrosis and tubular atrophy, glomerulosclerosis (global or segmental), mesangial hypercellularity, endocapillary proliferation, crescents, necrotizing segments, vascular sclerosis, parietal immune deposits (subendothelial) and abundant IgG or IgM codeposits.
Although many of these variables have demonstrated correlation with the prognosis in studies of populations of patients, do not have very much value when an individual case is analyzed.
See "Case 3" as an example of IgA nephropathy.
Schönlein-Henoch (S-H) purpura is a disease characterized by acute nephritis associated to purpuric skin lesions, mainly in lower extremities and buttocks, migratory arthritis, and gastrointestinal hemorrhage. It is also known as S-H syndrome, S-H nephritis, anaphylactoid purpura, and rheumatoid purpura. Named for the German physicians Johann Lukas Schönlein (1793-1864) and Eduard Heinrich Henoch (1820-1910). Some authors think that Schönlein's name should come first because he was the first person to recognize the condition (in 1837) (although the condition appears to have first been described by Heberden in 1801 [Jennette JC, et al. Heptinstall's Pathology of the Kidney. 6th ed. Wolters Kluwer Lippincott Williams & Wilkins, Philadelphia, 2007]). Henoch in 1868 reported the first case of a patient with colic, bloody diarrhea, painful joints, and a rash. The syndrome is considered a form of IgAN with prominent extrarrenal involvement: systemic vasculitis mediated by IgA rich immune complexes. The disease is more frequent in children, but it can affect patients of any age. The histopathologic alterations can be similar to those of IgAN, including their immunopathologic and ultrastructural appearance.
Severity of glomerular lesions is very variable, but frequently there is necrotizing segments, endocapillary proliferation, and crescents. All these severe lesions must be quantified in a biopsy in which this disease is diagnosed. The ample spectrum of glomerular changes is similar the that of lupus nephritis, and it is possible also to find a membranoproliferative pattern. The lesions may be variable in the time in a same patient. There are also diverse histopathologic classifications of the disease, with variable correlation with the prognosis; most important is to make a rigorous quantification of crescents, segmental or diffuse proliferative lesions, and segmental areas with necrosis or sclerosis.
By immunofluorescence and electron microscopy the findings may be similar to those of IgAN, but most frequently there are subendothelial and subepithelial deposits, mainly the first. In some cases parietal deposits predominate over those mesangials. Like in IgAN the predominant subclass in the deposits is IgA1.
In the skin lesions there are IgA deposits similar to the glomerular deposits.
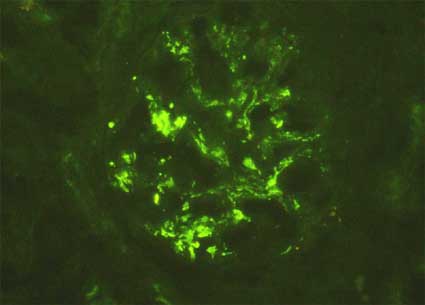
Figure 8. The immunopathologic characteristics in Schönlein-Henoch syndrome are usually similar to those of IgAN, as in this case of a 9-years-old patient with the disease; nevertheless, parietal immune deposits are more frequent, mainly subendothelial. (Immunofluorescence with anti-IgA antibodies marked with fluorescein, X400)..
The renal alterations mark the prognosis of the disease. Approximately 60% of patients present resolution of the disease, usually in the first 6 months; another 15 - 20% present smaller urinary abnormalities without deterioration of the renal function, and another 20 - 25% present progressive renal damage until terminal renal failure in 10 to 20 years. Findings of poor prognosis include macroscopic hematuria, a prolonged clinical course, severe proteinuria, arterial hypertension, percentage of crescents (> 50%) and, in some works, extension of the subendothelial deposits.
The overall outcome is good in most patients. All of the manifestations of active S-H purpura usually resolve spontaneously, although recurrent episodes of skin rash and hematuria may be seen. Among those with kidney involvement, only a minority have persistent disease. The kidney prognosis is excellent in most patients. However some patients will have persistent protein in their urine, high blood pressure, and renal insufficiency. It is estimated that S-H purpura accounts for approximately 3% of cases of end-stage kidney disease in children. Poor renal prognosis is more common among those with the nephrotic syndrome, renal insufficiency, and more advanced findings on biopsy. About 50% of patients have at least 1 recurrence. Patients <3 years-old usually have an improved prognosis. The prognosis is best for patients with minimal or no renal involvement at the onset of the illness (Dyne P, DeVore HK. Henoch-Schönlein Purpura. In: eMedicine, consulted August 23rd 2007. [Link: www.emedicine.com/emerg/topic767.htm]). Between 20 and 50 percent of children with HSP develop some kidney problems, but only 1% progress to total kidney failure. Progression to kidney failure may take as long as 10 years.
Recurrences are common, occurring in approximately one-third of patients. Since complete recovery is frequent, most patients receive no specific therapy. There is suggestive evidence that corticosteroids enhance the rate of resolution of the arthritis and abdominal pain, although they do not appear to prevent recurrent disease. Specific treatment is recommended in patients with marked proteinuria and/or impaired kidney function during the acute episode. A kidney biopsy can be performed to reveal the severity of the lesions which appears to be the best indicator of prognosis. Advanced disease, usually defined as crescentic nephritis, is treated with a regimen consisting of pulse intravenous methylprednisolone followed by oral prednisone. Other regimens include azathioprine, cyclophosphamide, and plasmapheresis, however, since spontaneous recovery is often observed in these patients, it remains unknown whether these regimens are superior to no or less aggressive therapy (Anonymus. Henoch-Schönlein Purpura. In: Vasculitis Foundation. Consulted August 23rd 2007. [Link: www.vasculitisfoundation.org/HenochSchonleinpurpura]).
Kidney transplantation can be performed in those patients who progress to end-stage kidney disease, although recurrent disease can occur. This appears to be more likely in patients with aggressive initial disease who progressed to end-stage kidney disease in less than three years after the onset of S-H purpura. Therefore it is recommended the transplantation to be delayed for 12-24 months after the disappearance of the rash. Some observations suggest that the risk of recurrent disease also may be higher in living-related donors.
In the treatment steroids or other immunosuppressors usually are used, but there is no universal agreement with respect to the optimal treatment. The disease can recur post-transplant (~53%), but usually like IgA deposition without deterioration of the function; recurrent clinical nephritis is detected in approximately 18% of cases; in ~9% of transplanted patients we will find loss of the graft as a result of the disease.
See Case 18 and Case 45 of our case series
Recent bibliography:
Homepage - - - Tutorial Index